Quality control can use fastQC. In DNA-seq data, we may find lots of overrepresented sequences from mitochondria and it is totally normal. Not like RNA-seq, we do not expect errors in GC content.
mkdir fastQC/ fastqc -o ./fastQC/ -t 7 reads.fq
for loop for slumn system
Example file:
GFP-1_L1_1.fq.gz
GFP-1_L1_2.fq.gz
FQ_DB={dir for fastq}
for i in $(ls $FQ_DB/* ); do SAMPLE=$(echo$i|sed 's=FQ/==;s/.fq.gz//') sed "s/Hi/$SAMPLE\_fQC/" Model.sh > script/$SAMPLE\_fQC.sh echo fastqc -o fastQC -t 7 $i >> script/$SAMPLE\_fQC.sh sbatch script/$SAMPLE\_fQC.sh done
Align to the Genome
Choose appropriate short reads tools for your data. I heard that bwa was more suitable for short reads align, so it is frequently used in the miRNA-Seq pipeline. Bowtie is faster and can do better on longer reads alignment. According to this, if your reads are short like 50~70bp, you can choose bwa. If your reads are paired from 150~300bp, bowtie2 would work better.
In the post from David, 2015; the best short-reads alignment tool is segemehl. Meanwhile, bwa was more sensitive than bowtie2. According to the Benchmark, the best tools for DNA-Seq is segemehl or BWA-MEM, the best tools for RNA-Seq is segemehl
In this Part, we will map all reads from the same directory to the reference genome by using bwa. Because programs would fail sometimes and we need to run them again. So, we can check and run only when the results don’t exist.
FQ_DB={Your Directory} DB={Your Reference Genome}
for SAMPLE in $(ls $FQ_DB/*L001*|awk -F/ '{print $NF}'| awk -F_ '{print $1"_"$2}'| sort |uniq); do if [ -f $SAMPLE.sorted.bam ]; then echo$SAMPLE is done else echo$SAMPLE can not be find sed "s/Hi/$SAMPLE/" Model.sh > script/$SAMPLE.sh echo cat $FQ_DB/$SAMPLE* \| bwa mem $DB - -t 64 \|samtools view -S -b -\|samtools sort \> $SAMPLE.sorted.bam >> script/$SAMPLE.sh echo samtools index $SAMPLE.sorted.bam >> script/$SAMPLE.sh sbatch script/$SAMPLE.sh fi done
## Check unexpected size of file rm $(du *.sorted.bam | awk '$1<=100'| awk '{print $2}') ## After removed failed files, we can excute the for loop again
for SAMPLE in $(ls $FQ_DB/*L001*|awk -F/ '{print $NF}'| awk -F_ '{print $1"_"$2}'| sort |uniq); do if [ -f $SAMPLE.sorted.bam ]; then echo$SAMPLE is done else echo$SAMPLE can not be find sed "s/Hi/$SAMPLE/" Model.sh > script/$SAMPLE.sh echo mv $SAMPLE.sorted.sam $SAMPLE.sorted.bam >> script/$SAMPLE.sh echo samtools index $SAMPLE.sorted.bam >> script/$SAMPLE.sh sbatch script/$SAMPLE.sh fi done
for i in $(ls Bam/*.bam); do SAMPLE=$(echo$i| sed 's=Bam/==;s/.sorted.bam//'); if [ -f Bam_stats/$SAMPLE.stats.csv ]; then echo$SAMPLE is done else sed "s/Hi/$SAMPLE\_BamStat/" Model.sh > script/$SAMPLE\_BamStat.sh echo samtools stats $i \> Bam_stats/$SAMPLE.stats.csv >> script/$SAMPLE\_BamStat.sh sbatch script/$SAMPLE\_BamStat.sh fi done
After jobs are done, we can grep the SN infor from all samples into one:
grep ^SN Bam_stats/*| sed 's/.stats.csv:SN//;s=Bam_stats/==' | awk -F"\t"'{OFS="\t"; print $1,$2,$3}'| sed 's/://' > SN_infor.csv
Negative insert sizes can happen when the first read is mapped to the reverse strand. we can change it by using awk '{print sqrt(\$0^2)}'
Reference: accio, 2020
It might be help in ChIP-seq data.
for i in $(ls Bam/SJ_*.bam); do SAMPLE=$(echo$i| sed 's=Bam/==;s/.sorted.bam//'); if [ -f Bam_stats/$SAMPLE.insert.csv ]; then echo$SAMPLE is done else sed "s/Hi/$SAMPLE\_BamStat/" Model.sh > script/$SAMPLE\_BamInser.sh echo samtools view -f66 $i \| cut -f9 \| awk "'{print sqrt(\$0^2)}'" \> Bam_stats/$SAMPLE.insert.csv >> script/$SAMPLE\_BamInser.sh bash script/$SAMPLE\_BamInser.sh & fi done
grep . Bam_stats/*.insert.csv| sed 's=Bam_stats/==;s/.insert.csv:/ /' > Insert.csv
Check the number of lines from each file and remove incorrect files.
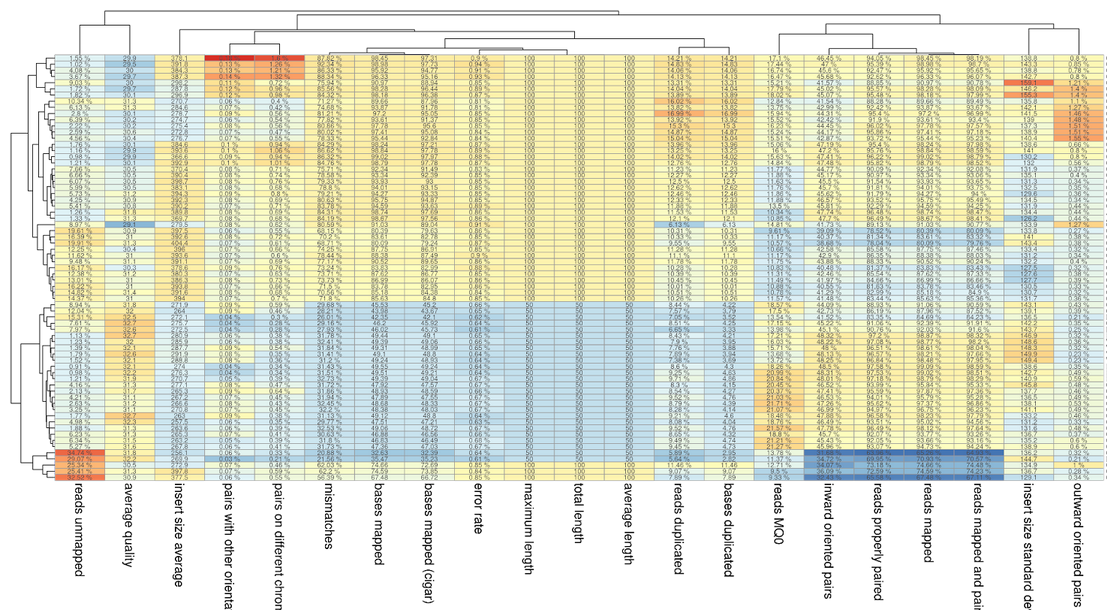
P <- pheatmap(as.data.frame(scale(t(TB_wS))), display_numbers = t(TB_w[row.names(TB_w) %in% row.names(TB_wS),]), fontsize_col= 15) as.ggplot(P) ggsave("img/Stat_Pheatmap.png", w= 20, h = 10.9)
Pheatmap
Align Depth Plot
Depth statistics are based on samtools: samtools depth -r UAS:0-10000 sorted.bam
mkdir Depth for i in $(ls Bam/*.bam); do SAMPLE=$(echo$i| sed 's=Bam/==;s/.sorted.bam//'); if [ -f Depth/$SAMPLE.csv ]; then echo$SAMPLE is done else sed "s/Hi/$SAMPLE/" Model.sh > script/Depth_$SAMPLE.sh echo samtools depth -r UAS:0-10000 $i \> Depth/$SAMPLE.csv >> script/Depth_$SAMPLE.sh sbatch script/Depth_$SAMPLE.sh fi
done
rm *.err *.out grep . Depth/*.csv| sed 's/.csv//;s/:/\t/;s=Depth/==' > Depth.csv
#!/bin/sh #SBATCH --qos=normal # Quality of Service #SBATCH --job-name=Hi # Job Name #SBATCH --time=1-0:00:00 # WallTime #SBATCH --mem=256000 #SBATCH --mail-user=123@qq.com #SBATCH --nodes=1 # Number of Nodes #SBATCH --ntasks-per-node=1 # Number of tasks (MPI processes) #SBATCH --cpus-per-task=1 # Number of threads per task (OMP threads) #SBATCH --output=Log/Hi.out ### File in which to store job output #SBATCH --error=Log/Hi.err ### File in which to store job error messages