Cell Ranger for scRNA-Seq Data Analysis
References
Cell Ranger is a software suite for analyzing single-cell RNA sequencing (scRNA-Seq) data, developed by 10x Genomics. It provides tools for processing raw sequencing data, performing quality control, and generating gene expression matrices.
It has detailed documentation available on the 10x Genomics website.
Basically, we only need to download the fasta and gtf files for the reference genome, and then run the cellranger mkgtf command with the appropriate parameters.
A very sample tutorial from 10x Genomics after you download the reference genome:
|
Why Filter???
Filtering the GTF file to include only protein-coding genes is a common practice in single-cell RNA-seq analysis. This is because the focus is typically on the expressed genes that are relevant for downstream analyses, such as clustering and differential expression. Non-coding genes, pseudogenes, and other non-protein-coding elements may not provide useful information for these analyses and can add noise to the data.
10x Genomics provides a detailed guide on the filter log of reference they provide. For human GRCh38, they using the following command:
"(protein_coding|protein_coding_LoF|lncRNA|
IG_C_gene|IG_D_gene|IG_J_gene|IG_LV_gene|IG_V_gene|
IG_V_pseudogene|IG_J_pseudogene|IG_C_pseudogene|
TR_C_gene|TR_D_gene|TR_J_gene|TR_V_gene|
TR_V_pseudogene|TR_J_pseudogene)"
So, double check your goal and then filter the GTF file accordingly.
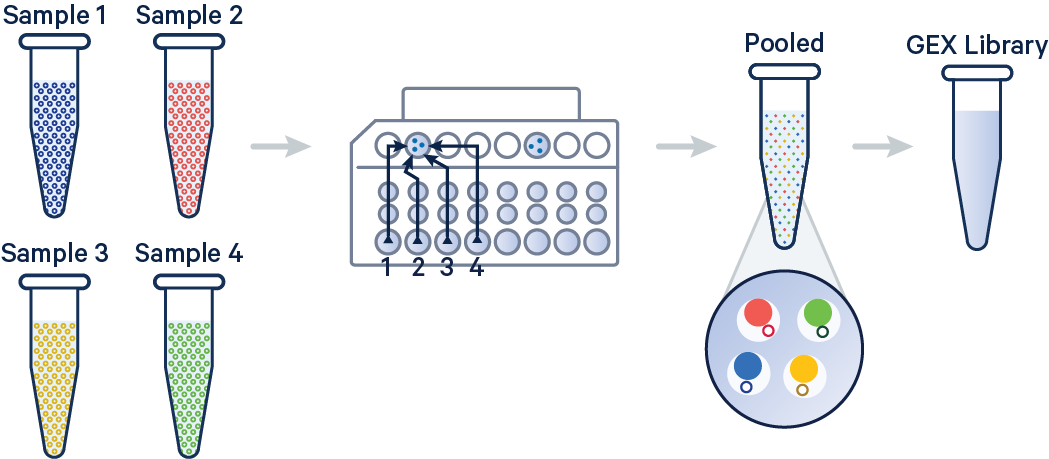
On Chip Multiplexing (OCM)
On Chip Multiplexing (OCM) is a feature in Cell Ranger that allows for the analysis of multiple samples in a single sequencing run. This is particularly useful for reducing costs and increasing throughput in single-cell RNA-seq experiments. More detailed pipeline can be found in the 10x Genomics documentation.
Example of data structure for OCM:
Project_Wu_VDJ ├── Sample_Rat_VDJ_S2_L004_I1_001.fastq.gz ├── Sample_Rat_VDJ_S2_L004_I2_001.fastq.gz ├── Sample_Rat_VDJ_S2_L004_R1_001.fastq.gz └── Sample_Rat_VDJ_S2_L004_R2_001.fastq.gz Project_Wu_GEX ├── Sample_Rat_GEX_S1_L004_I1_001.fastq.gz ├── Sample_Rat_GEX_S1_L004_I2_001.fastq.gz ├── Sample_Rat_GEX_S1_L004_R1_001.fastq.gz └── Sample_Rat_GEX_S1_L004_R2_001.fastq.gz
In this case, the Sample names are Sample_Rat_VDJ and Sample_Rat_GEX. This is the fastq_id we need to mention in the library section.
Error for sample_id
Technically, CellRanger looks at FASTQ files with this pattern: Sample_<sample_id>_Si and you just need to put the <sample_id> in the fastq_id field of the library section in the sample_sheet.csv file. But you can get error in some cases and you have to use Sample_<sample_id> format. This part waste me lots of time.
In this case, I have both VDJ and GEX data. By using multi would be very easy and convient to run the Cell Ranger pipeline.
|
In the [libraries] section, the VDJ-B is the feature type for VDJ data which means V(D)J library from B cells (heavy/light chains, BCR).
After preparing the sample_sheet.csv file, you can run the Cell Ranger pipeline with the following command:
|
Cell Ranger for scRNA-Seq Data Analysis
https://karobben.github.io/2025/07/09/Bioinfor/cellranger-scRNA/







