Understanding and Tackling Batch Effects in Single-Cell RNA-Seq Analysis
The Challenge of Batch Effects
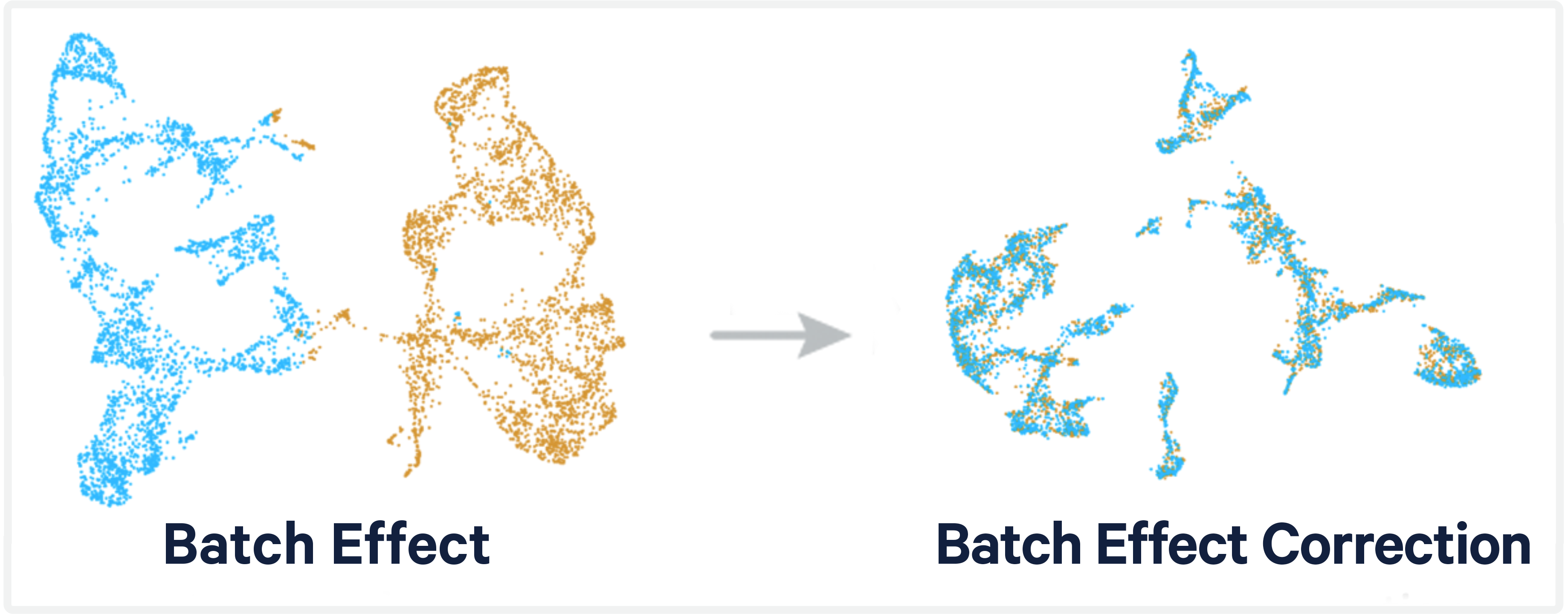
In the world of single-cell RNA sequencing (scRNA-seq), one of the most prevailing challenges is the presence of batch effects. These are technical non-biological variations that arise when samples are processed in separate runs or under slightly different conditions. If not accounted for, batch effects can overshadow true biological differences, leading to misinterpretations.
Why Remove Batch Effects?
Imagine studying the effects of a drug on cell populations, with samples processed both before and after treatment. If the ‘before’ samples were processed in one batch and the ‘after’ samples in another, any difference you observe might be due to this batch variation rather than the drug effect.
Removing batch effects ensures that:
- Biological variations are distinguishable from technical variations.
- Combined data from multiple batches can be analyzed together without biases.
- Results are consistent, reproducible, and truly reflective of biological phenomena.
Approaches to Counter Batch Effects
Several methods have been developed to correct for batch effects. Here, we’ll delve into three popular methods used within the Seurat package: Harmony, fastMNN, and SCTransform.
A Comparative Glimpse:
| Feature | Harmony | fastMNN (batchelor) | SCTransform |
|---|---|---|---|
| Method | Operates in PCA space to adjust principal components. | Uses Mutual Nearest Neighbors to align cells. | Regresses out unwanted variation and stabilizes variance. |
| Use Cases | Integrating datasets from different conditions, protocols, or platforms. | Integrating datasets with batch effects. Faster than original MNN. | Alternative to traditional normalization in Seurat. Prepares data for analysis. |
| Advantages | Preserves biological structures well; robust to complex batch effects. | Designed for scRNA-seq batch correction; faster than original MNN; corrects severe batch effects. | Stabilizes variance; can handle large datasets efficiently. |
| Disadvantages | Can be computationally intensive with large datasets. | Requires a good selection of mutual nearest neighbors. | Not specifically a batch correction method; often combined with other methods. |
Deep Dive into the Methods:
-
Harmony: Ideal for integrating multiple scRNA-seq datasets. Harmony adjusts the principal components of the data such that inter-batch differences are minimized while retaining biological variation.
-
fastMNN (from the batchelor package): This method identifies mutual nearest neighbors between batches, assuming that these pairs of cells represent the same cell type across batches. It’s an optimized and faster variant of the original MNN algorithm.
-
SCTransform: More than a batch correction technique, SCTransform is a robust normalization method. It prepares the data for downstream analysis, including batch correction, by regressing out unwanted sources of variation.
Final Thoughts:
Batch effect correction is a crucial step in scRNA-seq data processing. By choosing the right method tailored to your dataset’s needs, you can unveil genuine biological insights without being misdirected by technical noises. Always visualize and interpret results at each step, ensuring that biological variation remains the highlight of your study.
In Action
Seurat offers several methods to correct for batch effects, ensuring that variations across batches don’t obscure the biological signals you’re interested in. Here’s a step-by-step guide to remove batch effects using Seurat:
-
Install and Load Seurat:
First, ensure you’ve installed and loaded the Seurat package.
install.packages('Seurat')
library(Seurat) -
Data Integration (Using Harmony, SCTransform, and others):
Seurat offers various integration methods like Harmony, fastMNN, and SCTransform. Here, I’ll showcase using
SCTransformfollowed byRunPCAandHarmony:# List of Seurat Objects from different batches
seurat_list <- list(batch1_seurat, batch2_seurat, ...)
# SCTransform normalization
seurat_list <- lapply(seurat_list, function(x) {
x <- SCTransform(x, verbose = FALSE)
})
# Identify anchors for integration
anchors <- FindIntegrationAnchors(object.list = seurat_list, normalization.method = "SCT",
anchor.features = 2000, verbose = FALSE)
# Integrate data
integrated_data <- IntegrateData(anchorset = anchors, normalization.method = "SCT", verbose = FALSE)If you prefer using
Harmonyspecifically:library(harmony)
integrated_data <- RunPCA(integrated_data, features = VariableFeatures(object = integrated_data))
integrated_data <- RunHarmony(integrated_data, group.by.vars = "batch") -
Scale and Run Linear Dimensional Reduction:
integrated_data <- ScaleData(integrated_data, verbose = FALSE)
integrated_data <- RunPCA(integrated_data, verbose = FALSE) -
Cluster and Visualize Integrated Data:
Using UMAP or t-SNE to visualize the integrated data is a great way to confirm if the batch effects have been mitigated.
integrated_data <- RunUMAP(integrated_data, reduction = "harmony", dims = 1:30)
DimPlot(integrated_data, group.by = "batch") -
Optional - Regression of unwanted sources of variation:
If you are aware of specific unwanted sources of variation, you can regress them out using the
vars.to.regressparameter in theScaleDatafunction.seurat_object <- ScaleData(seurat_object, vars.to.regress = "batch")
Remember, the removal of batch effects should be done carefully. Overcorrection might lead to loss of genuine biological variation. It’s always a good practice to visualize and interpret the results at each step, ensuring the intended correction.
Lastly, Seurat’s integration methods and their parameters might evolve with time, so always consult the latest Seurat documentation or vignettes to ensure you’re using the most effective and up-to-date approaches.
Harmony, fastMNN, and SCTransform are all methods to handle batch effects and data normalization in single-cell RNA-seq data, but they operate based on different principles and have distinct purposes.
In summary:
- Harmony and fastMNN are batch-correction methods that work to align datasets from different batches or conditions.
- SCTransform is a normalization method that prepares the data for further analysis, including batch correction.
When dealing with batch effects, researchers often first normalize the data using SCTransform and then apply batch correction methods like Harmony or fastMNN to the transformed data. This two-step approach ensures that the data is both normalized and free of batch effects, making downstream analyses like clustering and differential expression more reliable.
Other tutorial:
Understanding and Tackling Batch Effects in Single-Cell RNA-Seq Analysis







