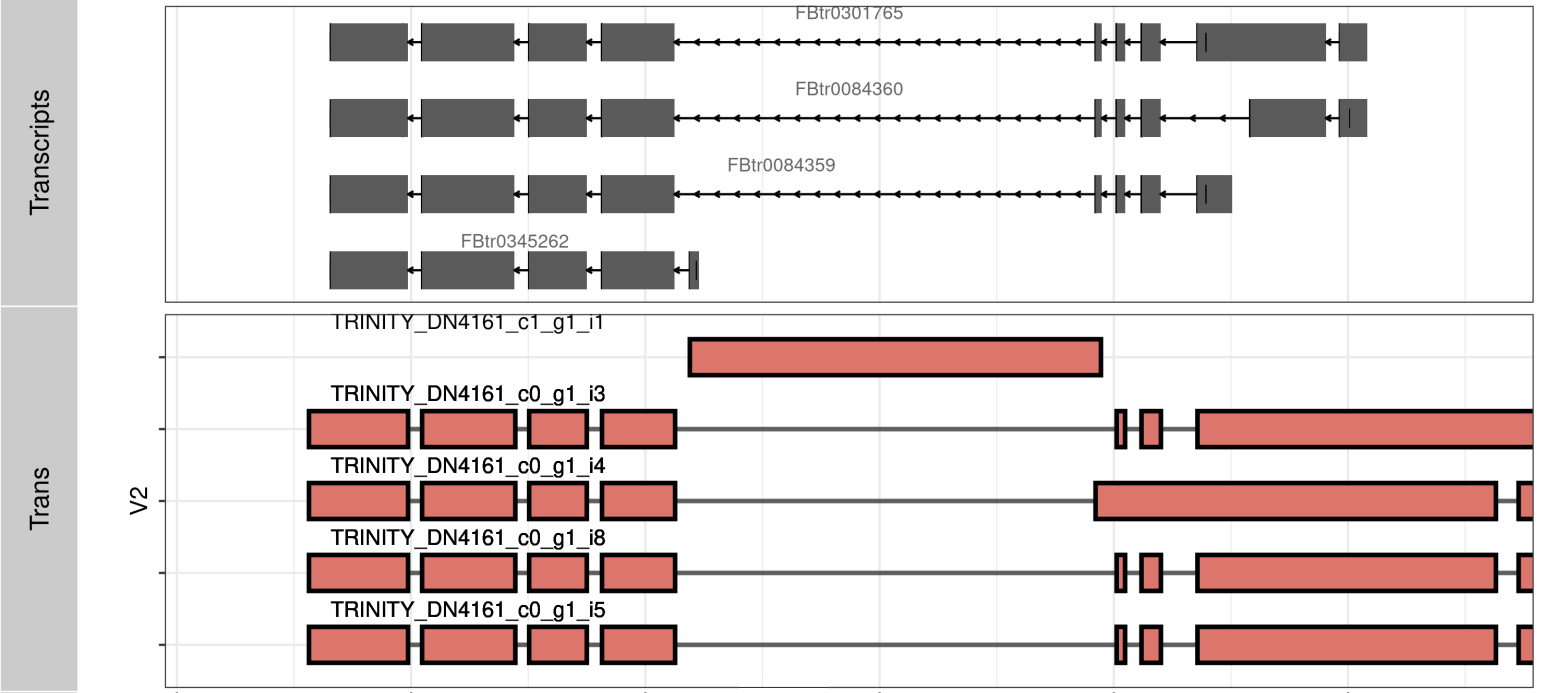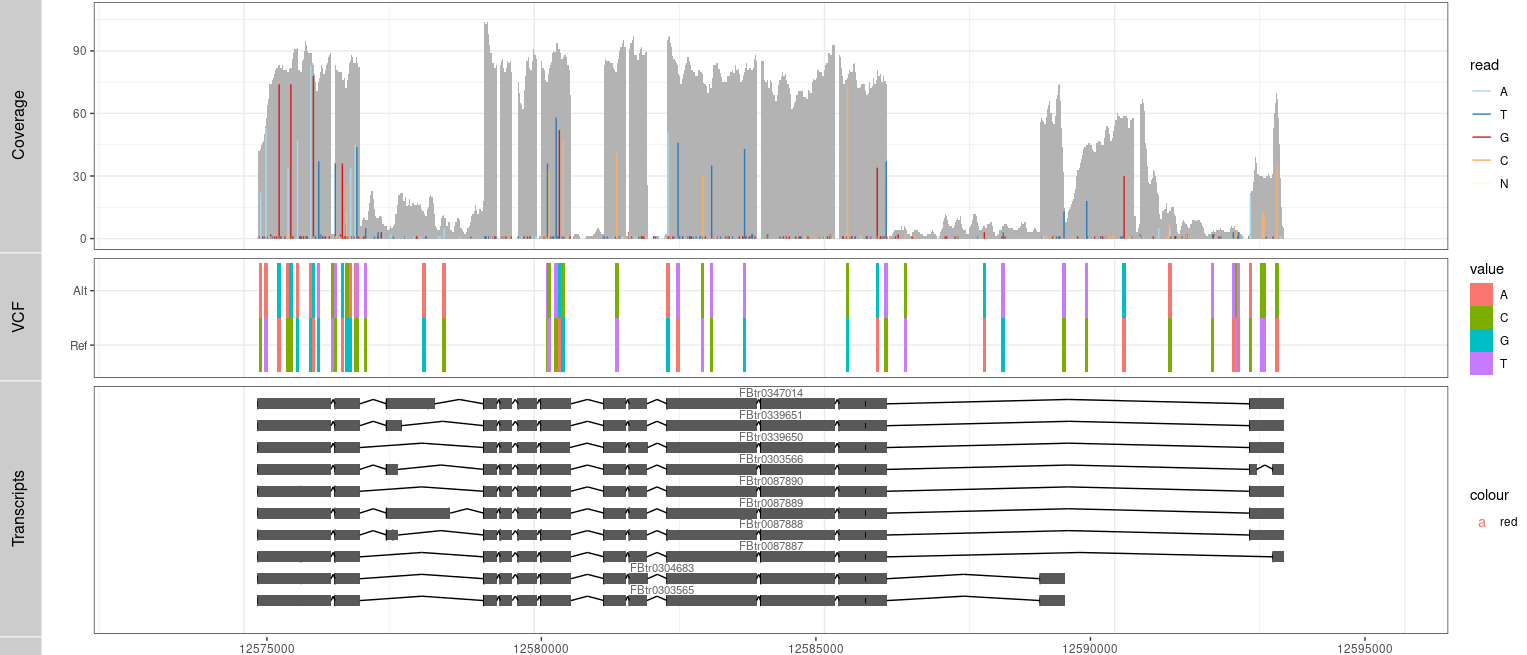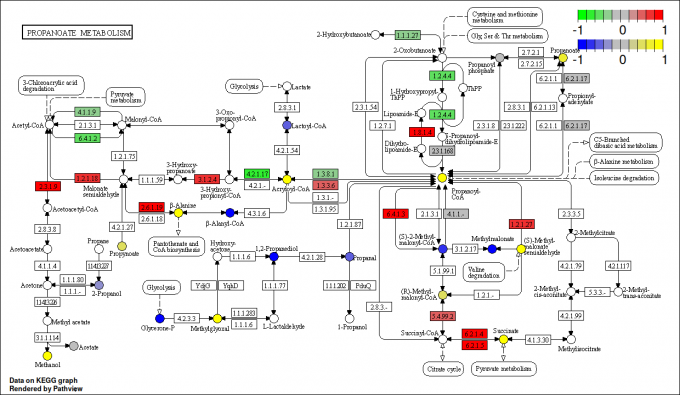
KEGG API
Learning KEGG API is important for accessing and integrating biological pathway data, enabling researchers to analyze and visualize complex biological systems, identify new potential drug targets, and explore the relationships between genes, proteins, and metabolic pathways. Who sad this?
Read more