Principles of Biochemistry 19 |Gluconeogenesis| Class Notes |HarvardX
Introduction
The brain will consume on average 120 grams of glucose per day. It is insufficient from the food.
Gluconeogenesis
Gluconeogenesis is not simply the reverse of glycolysis
- Limited reactions (3)
- Three metabolic valve enzymes (rate-limiting/Enzyme-limited)
- Those reactions are highly exergonic and irreversible.
- Substrate-limited reactions (7)
- Near equilibrium
- Reversible
Three bypass reactions
$$
Pyruvate + ATP + GTP + HCO_ 3 ^- \rightleftharpoons PEP + ADP + GDP + P_ i + CO_ 2
$$
$\Delta G’^ {\circ} = 0.9\ kJ/mol$
$[PEP]$ low
$\Delta G = -25\ kJ/mol$
The reaction is spontaneous and irreversible
- Pyruvate $\to$ PEP
In mitochondria: $Alanine \to Pyruvate$ - Pyruvate carboxylase
- Pyruvate + ATP $\overset{biotin}{\longrightarrow}$ Oxalocetate + ADP (coenzyme: Biotin)
- Oxalocetate + NADH $\overset{Malate\ dehydrogenase}{\rightleftharpoons}$ Malate + NAD+
(The change of free energy is nearly zero)
2.1. {Malate}Mitochondria $\overset{alpha-ketoglutarate\ transporter}{\longrightarrow}$ {Malate}Cytosol - Malate + NAD+ $\to$ Oxaloacetate + NADH
- Oxaloacetate + GTP $\overset{PEP\ carboxykinase}{\rightleftharpoons}$ PEP + GDP
Glyceraldehyde 3-phosphate NAD+ $\rightleftharpoons$ 1,3-Bisphosphoglycerate + NADH
Source of NADH
$$
[\frac{NADH}{NAD^ +}]_ {cyt} << [\frac{NADH}{NAD^ +}]_ {min}
$$
NADH is not permeable to the membrane of mitochondria.
So, the NADH does not come from mitochondria.
$Lactate + NAD^ + \to Pyruvate NADH$
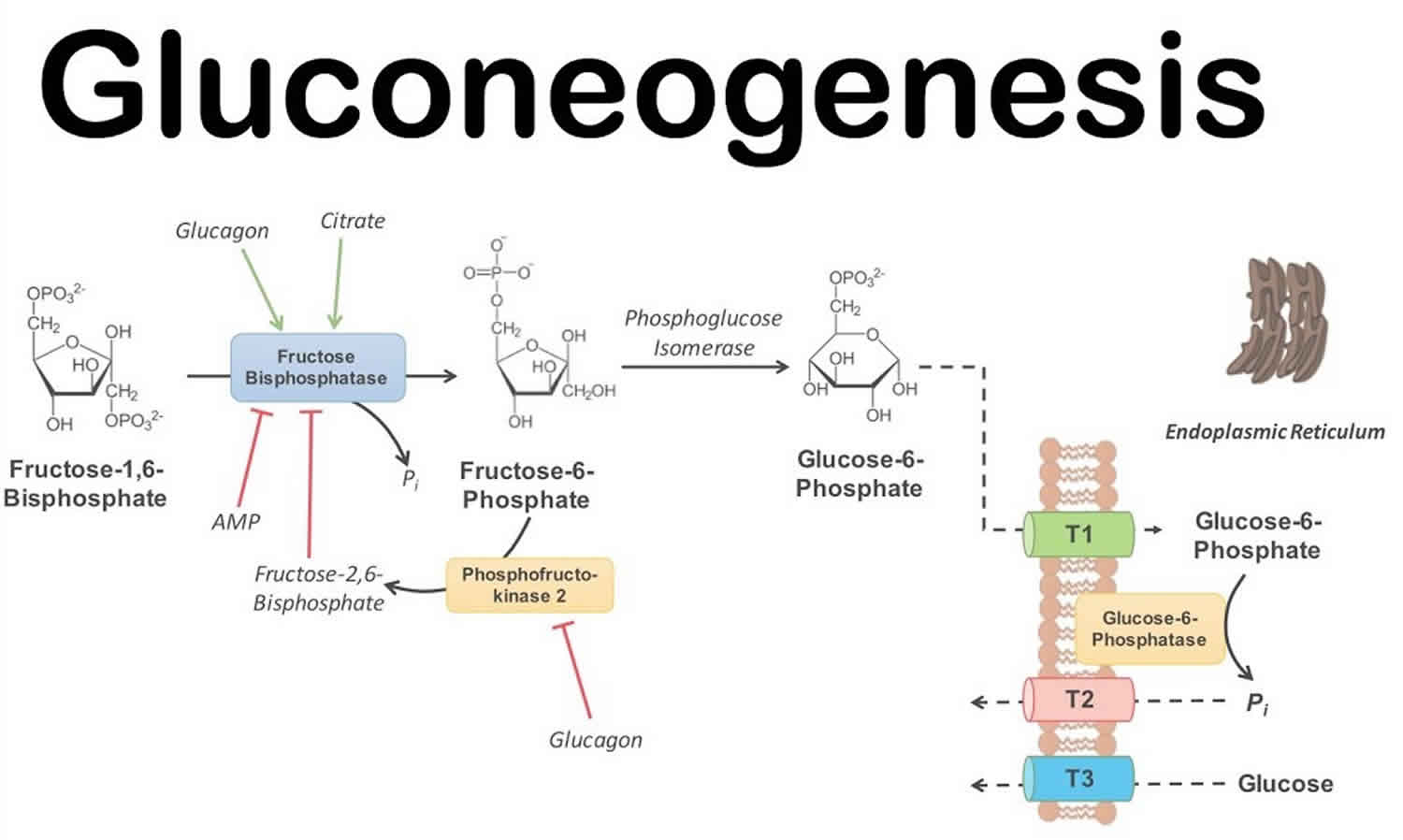
The second bypass reaction
$$
Fru-1,6-biphophotate \overset{Fructose\ 1,6\ biphosphatase}{\longrightarrow} Fru-6-phosphate
\
$$
$$
\Delta G’^ {\circ} = - 16.3kJ/mol
$$
The third bypass reaction
$$
Glucose-6-P \overset{Glucose\ 6\ biphosphatase}{\longrightarrow} Glucose
$$
$$
\Delta G’^ {\circ} = -13.8kJ/mol
$$
Free energy
| Glycolysis | Gluconeogenesis |
|---|---|
| -63 kJ/mol | -16 kJ/mol |
Blood glucose regulation
The liver:
- Balances glycolysis and Gluconeogenesis
- Controls glycogen synthesis and degradation
Glycogen metabolism is regulated by hormones.
Low blood glucose
As a result, the glycogen synthesis was inactivated.
High blood glucose
GSK is the inactivator of the Glycogen synthase. Without the inactivator, the activation of glycogen synthase was increased.
PP1
PP1 is a part of a complex called Glycogen synthase phosphatase which contains Glycogen-targeting regulatory subunit. It’s a member of the family of glycogen-targeting regulatory subunits and could recruit the glycogen synthase phosphatase to glycogen particles.
In high blood glucose level:
- PP1 dephosphorylates both enzymes (Glycogen synthase and Glycogen phosphorylase), so the glycogen synthase becomes activated, and the glycogen phosphorylase is now inactive.
- Inactivate a protein called phosphorylase kinase and inhibit glycogen breakdown
- Insulin would decrease the concentration of cAMP which responsible to activate PKA. (PKA could prohibit the function of PP1)
(Glycogen phosphorylase: breakdown the glycogen)
In low a concentration of blood glucose, PKA inhibits the PP1.
==In low blood glucose level
Glycogen phosphorylase would bind to the Glycogen synthase phosphatase which inhibited the PP1. So, glycogen synthesis is inhibited.
Glycogen-targeting regulatory subunit has two kinds of isoform: GL in the muscle.
regulation in muscle
low blood glucose
- PKA cold phosphorylates GM. Upon phosphorylation, PP1 dissociates from GM which could reduce the catalytic efficiency of PP1. (Substrate is recognized and recruited by GM).
- PKA also phosphorylates the inhibitor of the PP1 and then binds it to the PP1 to inhibit the activity of PP1.
delay effect on synthase
- No additional Glucose: Glycogen phosphorylase is in an R state
- Added Glucose: turn to T state, PP1GL complex is released in now an active form.
- T state of Glycogen phosphorylase is more accessible for PP1GL complex
- PP1GL complex could now quickly bind the glycogen synthase and stimulation the glycogen synthesis
Diabetes
Diabetes Mellitus:
- Elevated levels of blood sugar
- Deficiency in insulin activity
Two main groups of diabetes:
- Type I
- juvenile-onset diabetes (age of 25)
- Genetic, autoimmune, environmental
- Reduced insulin secretion
- $\beta$-cell loss
- Hard to prevent
- Type II
- Adult-onset (40 to 60 years)
- Genetic and environmental factors
- Resistance towards insulin
- Eventual $\beta$-cell loss and reduced insulin secretion
- relatively easy to prevent
- Gestational (pregnant woman)
- Temporary insulin resistance
- Relapse may occur (to Type II diabetes)
Common complication:
- Hyperglycemia
- Organ damage (eyes, kidneys, and nerves.)
Controlling Type II Diabetes:
- Long-term and continuous regulation of blood sugar
- Strict dietary control
Type I diabetes (Insulin-dependent diabetes)
Less than 10% of the diabetics
- sudden onset
- Immune system attacks $\beta$-cells
Causes:
- Viral infections initiate autoimmunity
- Genetic factors contribute
Severe $\beta$-cell loss = insufficient insulin secretion:
- undernourished
- underweight
- ketone body production
- metabolic acidosis
- lower the blood pH
Genetic factors: TYK2
TYK2 is the protein of which associate with interferon receptor, which presents INF $\alpha/\beta$ and recognized by the phagocyte.
The ineffectiveness of TYK2 loss of function increases the protection against type 1 diabetes by decreasing the antiviral and apoptotic responses.
PTPN2 could prevent the down-regulation of the antiviral in apoptotic activities.
Lipolysis
Inhibition of hormone-sensitive lipase: HSL
- HSL could convert diacylglycerol into monoacylglycerol.
Triacylglycerol (TAG) synthesis: - Conversion of glucose to glycerol 3-P in liver and adipocytes.
- Increased TAG synthesis in liver and adipocytes
- Increased TAG transport from liver to adipocytes
- Upregulation of lipoprotein lipase gene
Most tissues: - Increased amino acid uptake
- Activation of translation initiation factors
- Activation of protein synthesis
Hyperglycemia
High glucose in blood, very low glucose in the cell
- High fasting blood glucose > 126 mg/dL
- excess of glucose in the urine
- Osmotic pressure
- Exessive urination, polyuria, and dehydration
- Excessive thirst, polydipsia
- Cell stress
- Tissue Damage: Exp, oxidative damage in $\beta$-cell
Tissue starvation
- Polyphagia: (feel constantly hungry)
- Glyconeogenesis
- Hyperglycemia
- Degrade lipids to fatty acids
- adipose tissue wasting
- ketone bodies synthesis
- Low blood pH and ketoacidosis
Hypertriacylglycerolemia
Excessive very-low-density lipoproteins due to low activity of lipoprotein lipase
Effect on insulin-insensitive tissues
- protein glycation
- small blood vessels
- retina
- red blood cells
Treatment for Type I Diabetes
It is caused by loss of insulin secretion because of loss of $\beta$-cell.
The obvious treatment is the administration of purified insulin.
- Subcutaneous injection (fat layer)
- Other injection sites (the part rich in adipose tissues)
- abdomen
- top of your thigh
- back of your arm
- pump-assisted infusion
It should avoid quick absorption in order to prevent from frequent injection
Exogenous insulin administration
| Standard | intensive | Healthy | |
|---|---|---|---|
| blood glucose level | ~225mg/dL | ~150mg/dL | ~100mg/dL |
| glycated Hb levels | ~8-9% | ~7% | < 6% |
Side effect:
It was caused by the time and dose of the insulin injected.
Less or more could cause damages:
To many:
- blood glucose levels < 100mg/dL
- Hypoglycemia
- Children < 8 yrs: brain development
- Elderly: heart attacks and stroke
Type II diabetes
Type II diabetes is characterized by the cell loss responsible for the circulating insulin.
Glucose uptake
As a result, our body controls the level of blood glucose by dynamically manipulating the number of Glucose pumps (GLUT-4) on the surface of receptor cells which responsible for the circulating insulin.
Insulin Receptor
- Tyrosine phosphorylation of the receptor and IRS-1 recruitment
- $\overset{phosphatase}{\longrightarrow}$ Insulin Receptor inactivation
- $\overset{proteosome}{\longleftarrow}$ Receptor and IRS-1 degradation
- IRS-1 phosphorylation and PI3K activation
- $\overset{S/T Kinase}{\longrightarrow}$ IRS-1 inactivation
- Conversion of PIP2 into PIP3
- $\overset{phosphatase}{\longrightarrow}$ Conversion of PIP2 into PIP3
- Inactive PDK1 and Akt
- Decreased glucose uptake and glycogen synthesis
- $\overset{phosphatase}{\longrightarrow}$ Conversion of PIP2 into PIP3
- Activation of PDK1 and Akt
- Increased glucose uptake and glycogen synthesis
Insulin resistance
adipocytes lead to chronic inflammation
- Adipocyte secretion of anti-inflammatory molecules decreases and adipocyte’s secretion of pro-inflammatory molecules increases.
- Exp: MCP-1, a chemical that attracts macrophages.
- Glucose transport inside muscle is still normal
- Microphages infiltrated in adipose tissue secrete TNF-alpha
- Result: increase lipolysis followed by increased fatty acid export to tissue, including muscle.
- excessive fatty acid in tissue in ectopic fat deposition and interference with the targeting of glucose transporter at the surface of cells.
- The consequence is the progression toward full insulin resistance.
One hypothesis: insulin overproduction
Hyperglycemia could trigger a large amount of insulin production. Proinsulin is heavily produced in the inner cellular ER and Golgi system. Over synthesis of this could damage the function of the ER and Golgi, which failed to remove the C-protein and produce misfolded insulin. Accumulation of misfolded insulin triggered apoptosis.
Conclusion: Obesity is a risk factor leading to ectopic fat deposition and ultimately to lipotoxicity and chronic inflammation which finally developed into insulin resistance and insulin deficiency.
Prevention
- Aerobic exercise, reduced caloric intake, healthy diet
- Lower glucose production by the liver
- Increase insulin production or sensitivity (e.g., sulfonylureas)
Function of sulfonylureas
Normal signaling pathway:
Principles of Biochemistry 19 |Gluconeogenesis| Class Notes |HarvardX
https://karobben.github.io/2021/06/07/LearnNotes/edx-biochm-19/







