Pheatmap
Pheatmap
Quick Start
Create a matrix
|
Test1 Test2 Test3 Test4 Test5 Test6 Test7 Test8 Test9 Test10
Gene1 3.113742 0.61097912 2.256865 -0.08655622 2.824332 -0.3824264 2.826347 -0.8726526 2.264322 1.9392831
Gene2 0.973717 0.03505342 2.681469 0.14051028 4.208013 -0.9295952 2.699840 -0.8473697 2.077007 0.2552222
Gene3 3.068895 -1.40479792 1.695945 -1.02215630 2.410066 -1.0977577 1.115832 2.4075234 3.305659 -0.4305648
Gene4 1.739518 0.43744627 3.438071 0.88019288 2.531289 0.5330686 3.914910 -1.5281725 3.853844 0.2501110
Gene5 3.500457 -0.76045729 3.161374 -1.09123751 3.486412 -2.8363874 3.362371 -1.3717895 3.585826 1.4036547
Gene6 3.385101 -0.45133918 3.537412 -2.23503436 1.783411 0.9475567 3.016503 0.4012510 1.886079 0.2387753
|
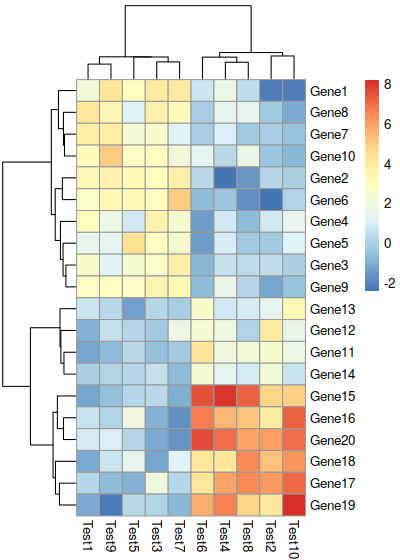
Arguments
Quick View:
pheatmap(mat, color = colorRampPalette(rev(brewer.pal(n = 7, name =
"RdYlBu")))(100), kmeans_k = NA, breaks = NA, border_color = "grey60",
cellwidth = NA, cellheight = NA, scale = "none", cluster_rows = TRUE,
cluster_cols = TRUE, clustering_distance_rows = "euclidean",
clustering_distance_cols = "euclidean", clustering_method = "complete",
clustering_callback = identity2, cutree_rows = NA, cutree_cols = NA,
treeheight_row = ifelse((class(cluster_rows) == "hclust") || cluster_rows,
50, 0), treeheight_col = ifelse((class(cluster_cols) == "hclust") ||
cluster_cols, 50, 0), legend = TRUE, legend_breaks = NA,
legend_labels = NA, annotation_row = NA, annotation_col = NA,
annotation = NA, annotation_colors = NA, annotation_legend = TRUE,
annotation_names_row = TRUE, annotation_names_col = TRUE,
drop_levels = TRUE, show_rownames = T, show_colnames = T, main = NA,
fontsize = 10, fontsize_row = fontsize, fontsize_col = fontsize,
angle_col = c("270", "0", "45", "90", "315"), display_numbers = F,
number_format = "%.2f", number_color = "grey30", fontsize_number = 0.8
* fontsize, gaps_row = NULL, gaps_col = NULL, labels_row = NULL,
labels_col = NULL, filename = NA, width = NA, height = NA,
silent = FALSE, na_col = "#DDDDDD", ...)
Color
|
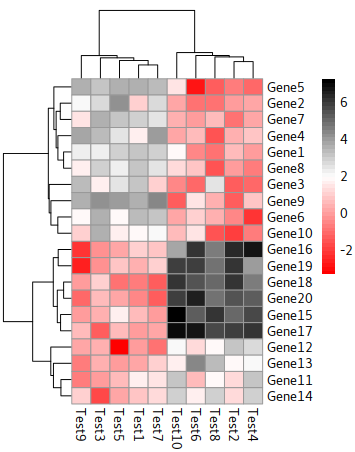
Dendrogram
|
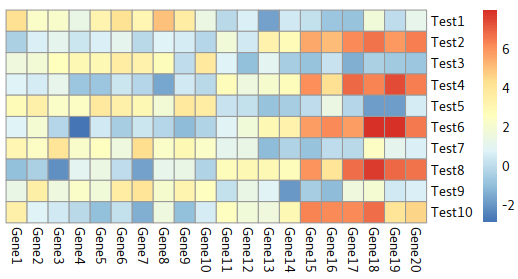
Disable dendrogram
|
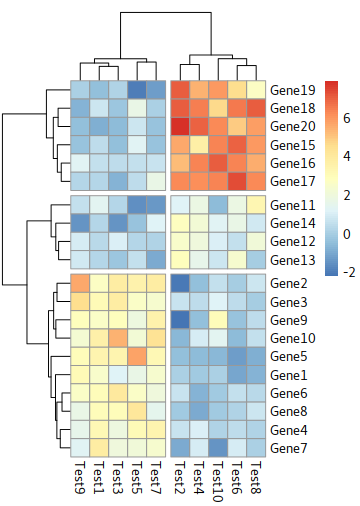
Cut trees
the parameters:
cutree_rows=int and cutree_cols=int
for example:
|
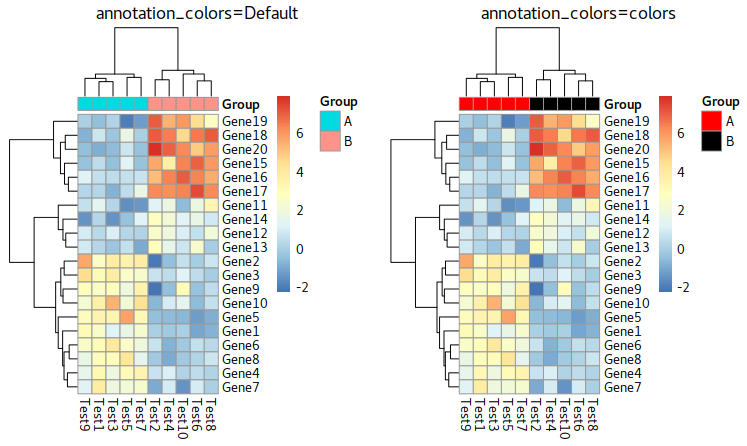
Group Annotation
In pheatmap, we use a matrix to store the annotaiton tags. An example you can see at below is Group which its rownames inhereted from colnames of test
Parameter about annotation
annotation_row = NA, annotation_col = NA, annotation = NA, annotation_colors = NA, annotation_legend = TRUE, annotation_names_row = TRUE, annotation_names_col = TRUE,
Example:
|
Group Test1 A Test2 B Test3 A Test4 B Test5 A
|
Two or more Layers of annotation
|
Group_2 Phy
Gene1 A 甲
Gene2 A 甲
Gene3 A 甲
Gene4 A 甲
Gene5 A 甲
Gene6 A 甲

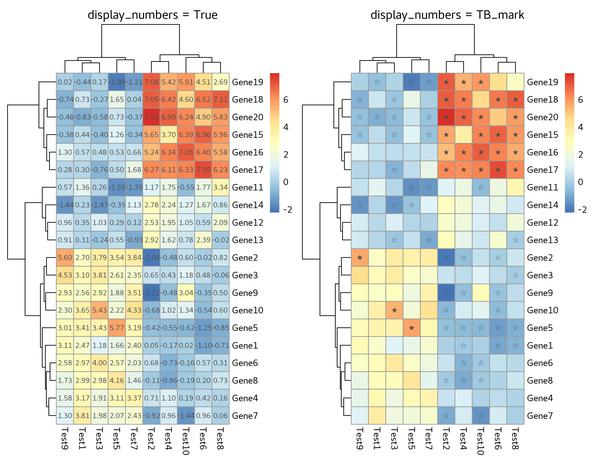
Display Numbers or Characters
display_numbers is the parameter we’d like to add.
|
Except for numbers, we can also adding characters,
for example:
|
Labels Annotation
|
Turn to ggplot
|
Heatmap for DEGs matrix
reference: Trinity
|
|
Parameters
mat:用来画热图的数据参数,一般是一个矩阵,数据是基因表达值,行代表基因,列代表样本。 color:表示颜色,用来画热图的颜色,可以自己定义,默认值为colorRampPalette(rev(brewer.pal(n = 7, name =”RdYlBu”)))(100),RdYlBu也就是Rd红色,Yi黄色,Bu蓝色的过度,则主调色为红黄蓝。 scale:是指对数值进行均一化处理,在基因表达量的数据中,有些基因表达量极低,有些基因表达量极高,因此把每个基因在不同处理和重复中的数据转换为平均值为0,方差为1的数据,可以看出每个基因在某个处理和重复中表达量是高还是低,一般选择做row均一化。 clustering_method:表示聚类方法,值可以是hclust的任何一种,如”ward.D”,”single”, “complete”, “average”, “mcquitty”, “median”, “centroid”, “ward.D2″。 cluster_rows:表示行是否聚类,值可以是FALSE或TRUE clustering_distance_rows:行距离度量的方法,如欧氏距离 cutree_rows:行聚类数 treeheight_row:行聚类树的高度,默认为50 gaps_row:对行进行分割,就不应对相应的行进行聚类 cluster_cols:表示列是否聚类,值可以是FALSE或TRUE clustering_distance_cols:列距离度量的方法 cutree_cols:列聚类数 treeheight_col:列聚类树的高度,默认为50 gaps_col:对列进行分割,就不应对相应的列进行聚类 legend:逻辑值,是否显示色度条,默认为T legend_breaks:显示多少个颜色数值段 legend_labels:对色度条上对应位置的字符进行修改 annotation_colors:对标签的颜色进行修改 annotation_legend:是否显示标签注释条 annotation_row:数据框格式,用来定义热图所在行的注释条 annotation_names_row:逻辑值,是否显示行标签名称 annotation_col:数据框格式,用来定义热图所在列的注释条 annotation_names_col:逻辑值,是否显示列标签名称 main:设置图的标题 fontsize:是设置所有除主图以外的标签的大小 number_color:字体的颜色 show_rownames:是否显示行名 fontsize_row:行名的字体大小 labels_row:X轴坐标名设置 show_colnames:是否显示列名 fontsize_col:列名的字体大小 labels_col:y轴坐标名设置 fontsize_number:小格子中数字大小 display_numbers:逻辑值,是否在小格子中显示数字 number_format:小格子中数字显示形式,但仅有在display_numbers=T时才能使用 na_col:设置小格子为缺失值时的颜色 cellwidth:表示每个小格子的宽度 cellheight:表示每个小格子的高度 filename:输出图画的文件名 width:输出图画的宽度 height:输出图画的高度
Reference: Davey1220 2018






