Short reads aligner compartment
Introduction
BWA
According to the Documentation
BWA is a software package for mapping low-divergent sequences against a large reference genome, such as the human genome. It consists of three algorithms: BWA-backtrack, BWA-SW and BWA-MEM.
- BWA-backtrack: It is designed for Illumina sequence reads up to 100bp,
- BWA-MEM: BWA-MEM and BWA-SW share similar features such as long-read support (70bp to 1Mbp) and split alignment. It is generally recommended for high-quality queries as it is faster and more accurate. It also has better performance than BWA-backtrack for 70-100bp Illumina reads.
QS
- There are three algorithms, which one should I choose?
For 70bp or longer Illumina, 454, Ion Torrent and Sanger reads, assembly contigs and BAC sequences, BWA-MEM is usually the preferred algorithm. For short sequences, BWA-backtrack may be better. BWA-SW may have better sensitivity when alignment gaps are frequent. - What is the tolerance of sequencing errors?
- Bwa-back is mainly designed for sequencing error rates below 2%.
- WA-SW and BWA-MEM both tolerate more errors given longer alignment. Simulation suggests that they may work well given 2% error for an 100bp alignment, 3% error for a 200bp, 5% for 500bp and 10% for 1000bp or longer alignment.
- Does BWA find chimeric reads?
Yes, both BWA-SW and BWA-MEM are able to find chimera. BWA usually reports one alignment for each read but may output two or more alignments if the read/contig is a chimera. - Does BWA work on reference sequences longer than 4GB in total?
Yes. Since 0.6.x, all BWA algorithms work with a genome with total length over 4GB. However, individual chromosome should not be longer than 2GB.
PS
chimeric reads: Chimeric reads occur when one sequencing read aligns to two distinct portions of the genome with little or no overlap. Chimeric reads are indicative of structural variation. Chimeric reads are also called split reads. After aligning with bwa mem, chimeric reads will have an SA tag as described on page 7 of the SAM format specification. To find them all you have to do is extract them using grep. donfreed, 2014. In the documentation, it mentioned that bwa aln is for find the SA reads.
Bowtie2
Bowtie 2 is an ultrafast and memory-efficient tool for aligning sequencing reads to long reference sequences. It is particularly good at aligning reads of about 50 up to 100s or 1,000s of characters, and particularly good at aligning to relatively long (e.g. mammalian) genomes. Bowtie 2 indexes the genome with an FM Index to keep its memory footprint small: for the human genome, its memory footprint is typically around 3.2 GB. Bowtie 2 supports gapped, local, and paired-end alignment modes.
What isn’t Bowtie 2?
Bowtie 2 is geared toward aligning relatively short sequencing reads to long genomes. That said, it handles arbitrarily small reference sequences (e.g. amplicons) and very long reads (i.e. upwards of 10s or 100s of kilobases), though it is slower in those settings. It is optimized for the read lengths and error modes yielded by typical Illumina sequencers.
Bowtie 2 does not support alignment of colorspace reads. (Bowtie 1 does.) Documentation
Bowtie2 could awareness splicing because it cut reads into even shorter before doing alignment.
PS
What is colorspace reads:
- Short answer: color space refers to the native format of ABI SoLID technology. Color space is translated to nucleotide, or base space (same thing) so that it can be understood.
- That technology is not growing in market share, so in the next few years it will become less common. ABI is putting most of their effort behind the Ion Torrent now. swbarnes2, 2012
Test Data
>1_Perfect_match TAATTCCCAAGATGAAGTTCCTGATCATCCTTGCCCTGGCTGTGGCCGCC >2_Intron_insertion TAATTCCCAAGATGAAGTTCCTGATTGCTGATCGATCGTAGCTAGCTAGCTAGCTAGCTAGCTACGTGCATCAGTCGATCAGTACGTCAGATGCTGTCGATCGTAGTCGATCGATGCTAGCTAGCTAGCTGCATAGTAGCTGCATGCTAGCTGCTAGCTCAGTAGCTCGTGCATGCATGCATCATCCTTGCCCTGGCTGTGGCCGCC >3_Intron_insertion TAATTCCCATGCTGATCGTGACTGCTGATCGATCGTGCTAGTCGATGCTCGTGCATGCTGCATGCTAGCTAGCTAGCTGACTGATCGTACGTCAGTGCATGCATGCTAGCTAGTAGCTAGCTAGCTAGCTCAGTCAGTCAGTCGATCGATGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGCTAGTCGATGCTAGCTAGCTAGCTCAGTCAGTAGCTCAGTCGAGCTGTGTGTGCTAGCACTACGTGTCGATGTGTCAGTAGATGAAGTTCCTGATCATCCTTGCCCTGGCTGTGGCCGCC >4_Cross_Chrm_L TAATTCCCAAGATGAAGTTCCTGATTGCTGATCGATCGTAGCTAGCTAGCTAGCTAGCTAGCTACGTGCATCAGTCGATCAGTACGTCAG >5_Cross_Chrm_R ATGCTGTCGATCGTAGTCGATCGATGCTAGCTAGCTAGCTGCATAGTAGCTGCATGCTAGCTGCTAGCTCAGTAGCTCGTGCATGCATGCATCATCCTTGCCCTGGCTGTGGCCGCC >6_Small_deletion_3 TAATTCCCAAGATGAAGTTCCTATCCTTGCCCTGGCTGTGGCCGCC >6_deletion_10 TAATTCCCAAGATGAAGTTCTTGCCCTGGCTGTGGCCGCC >6_deletion_20 TAATTCCCAAGATGAAGTTCTGTGGCCGCC >7_Samll_insertion_3 TAATTCCCAAGATGAAGTTCCTGATGATCATCCTTGCCCTGGCTGTGGCCGCC >8_SNP TAATTCCCAAGATGAAGTTCCTGATTATCCTTGCCCTGGCTGTGGCCGCC
Build reference DB (index)
|
Aligning
|
Results
Format of sam file:
A00327:224:HW2JVDRXY:1:1101:1253:1000 0 1_Perfect_match 1 60 50M * 0 0 TAATTCCCAAGATGAAGTTCCTGATCATCCTTGCCCTGGCTGTGGCCGCC #FFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFFF NM:i:0 MD:Z:50 AS:i:50 XS:i:0
| Columns | Abbr. | Exp | Describe |
|---|---|---|---|
| 1 | QNAME | A003… | Query (pair) NAME |
| 2 | FLAG | 0 | bitwise FLAG |
| 3 | RNAME | 1_Perfect_match | Reference sequence NAME |
| 4 | POS | 1 | 1-based leftmost POSition/coordinate of clipped sequence |
| 5 | MAPQ | 60 | MAPping Quality (Phred-scaled) |
| 6 | CIAGR | 50M | extended CIGAR string |
| 7 | MRNM | * | Mate Reference sequence NaMe (‘=’ if same as RNAME) |
| 8 | MPOS | 0 | 1-based Mate POSistion |
| 9 | ISIZE | 0 | Inferred insert SIZE |
| 10 | SEQ | TAAT… | query SEQuence on the same strand as the reference |
| 11 | QUAL | FFFF… | query QUALity (ASCII-33 gives the Phred base quality) |
| 12 | OPT | NM:i:0 MD:Z:50 AS:i:50 XS:i:0 | variable OPTional fields in the format TAG:VTYPE:VALUE |
| Reference | bowtie2 | bowtie | bwa |
|---|---|---|---|
| 1_Perfect_match | 1_Perfect_match | 1_Perfect_match | 1_Perfect_match |
| 2_Intron_insertion | * | NA | * |
| 3_Intron_insertion | * | NA | 3_Intron_insertion |
| 4_Cross_Chrm_L | * | NA | * |
| 5_Cross_Chrm_R | * | NA | * |
| 6_deletion_10 | * | NA | * |
| 6_deletion_20 | * | NA | * |
| 6_Small_deletion_3 | 6_Small_deletion_3 | NA | 6_Small_deletion_3 |
| 7_Samll_insertion_3 | 7_Samll_insertion_3 | NA | 7_Samll_insertion_3 |
| 8_SNP | 8_SNP | 8_SNP | 8_SNP |
| Ref | 1_Perfect_match | 1_Perfect_match | 1_Perfect_match |
Aligning Reads with Multiple Hits Using Bowtie2
By default, Bowtie2 reports only one alignment (the best one) for each read. However, you can configure it to report multiple alignments using the -k or -a options:
- k
: Report up to alignments per read (useful for getting multiple alignments if they exist). - a: Report all valid alignments (this can produce a large output if there are many alignments).
Example command to report up to 5 alignments per read:
|
TopHat
Unlike BWA and bowtie2, TopHat is a intron awareness aligner. It not only automatically split mapped and unmapped bam, but also given variation results like ‘junction’, ‘deletion’, and ‘insertion’ bed files. But the align performance of TopHat2 is not optimistic from some comparison.
column name of 'junction.bed':
[seqname] [start] [end] [id] [score] [strand] [thickStart] [thickEnd] [r,g,b] [block_count] [block_sizes] [block_locations]
- "seqname": chromosome
- "start": is the start position of the leftmost read that contains the junction.
- "end": is the end position of the rightmost read that contains the junction.
- "id": is the junctions id, e.g. JUNC0001
- "score": is the number of reads that contain the junction.
- "strand": is either + or -.
- "thickStart" and
- "thickEnd": don't seem to have any effect on display for a junctions track. TopHat sets them as equal to start and end respectively.
- "r.g.b": "r","g" and "b" are the red, green, and blue values. They affect the colour of the display.
- "block_count": The block_count will always be 2. The two blocks specify the regions on either side of the junction.
- "block_sizes": "block_sizes" tells you how large each region is.
- "block_locations": it tells you, relative to the "start" being 0, where the two blocks occur. Therefore, the first block_location will always be zero. from: Alex124, 2012
basic use:
|
More infor for bed fromat: UCSC
Hisat2
|
Others
Ryan Musich, et al using 48 fubgi’s RNA-seq samples, Erysiphe necator, to compare the results of aligners bowtie2, bwa, hisat2, Mummer4, star, and tophat2. They found that bowtie2 and bwa have less unmapped reads. TopHat2 is the worst which has arround 80% unmapped reads. Bowtie2 and bwa have a similar positive result but bwa consume much less time.
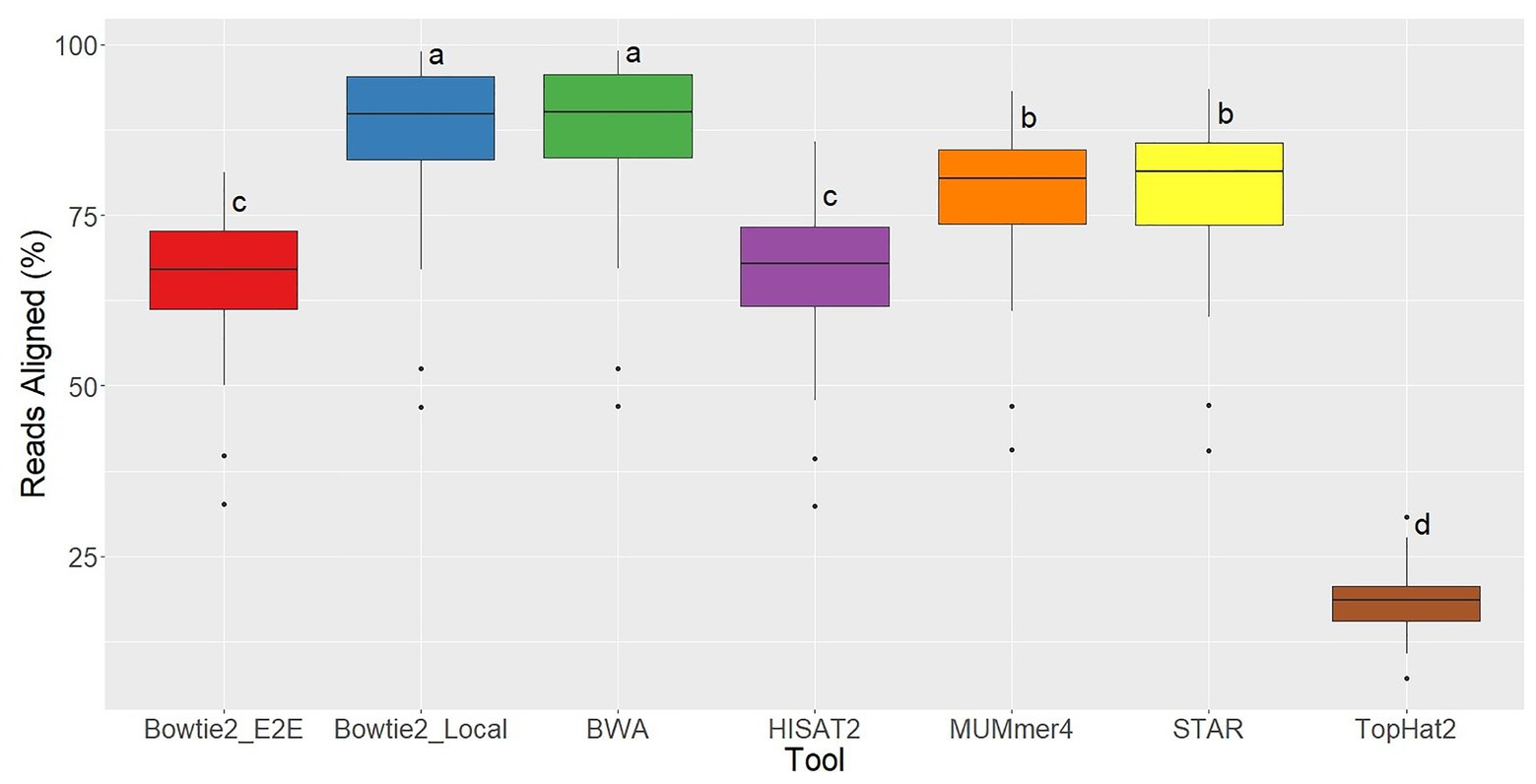 |
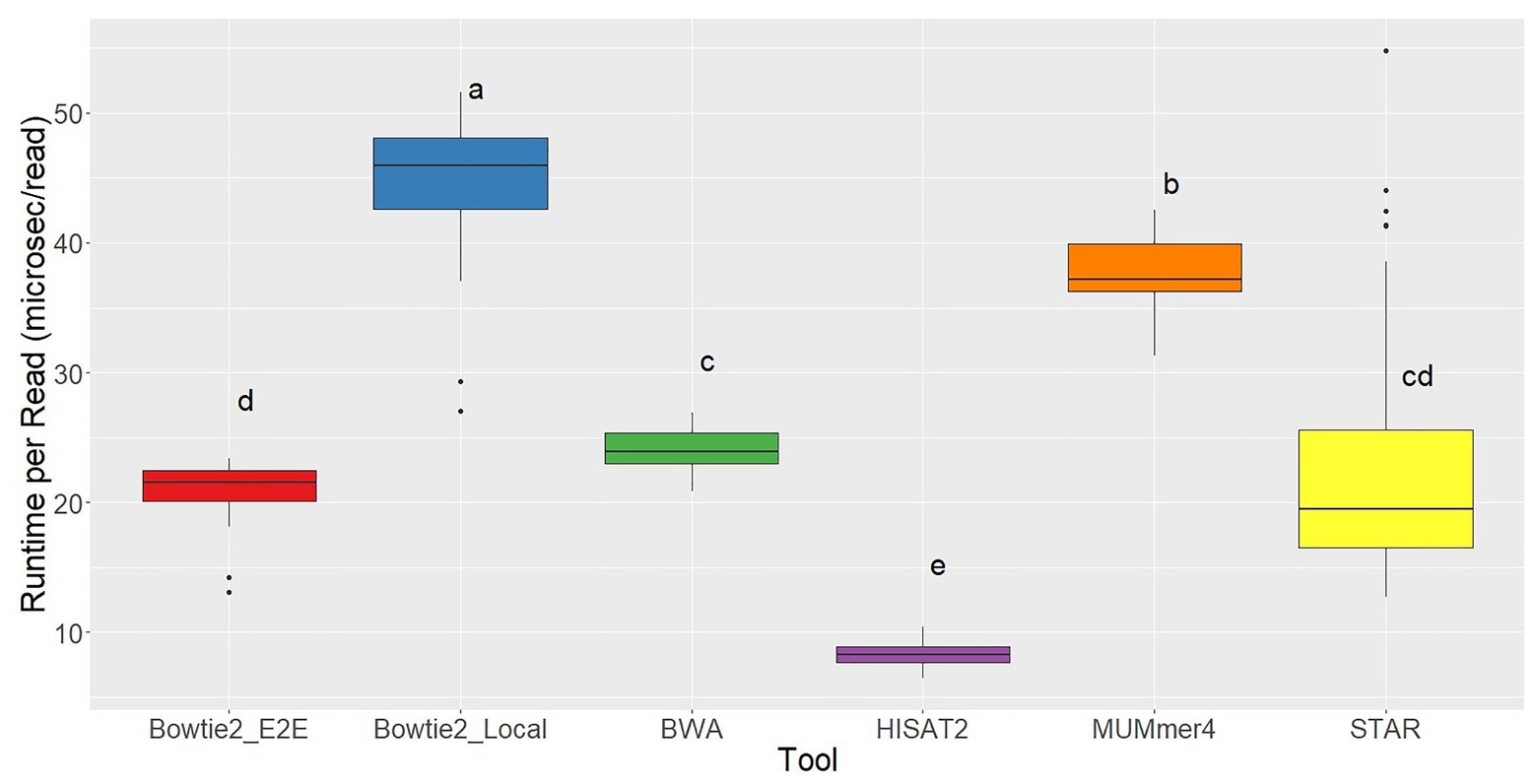 |
|---|
Grzegorz M. Boratyn, et al. developed a new tool Magic-BLAST for improving the accuracy of RNA-seq reads aligning. They compared the
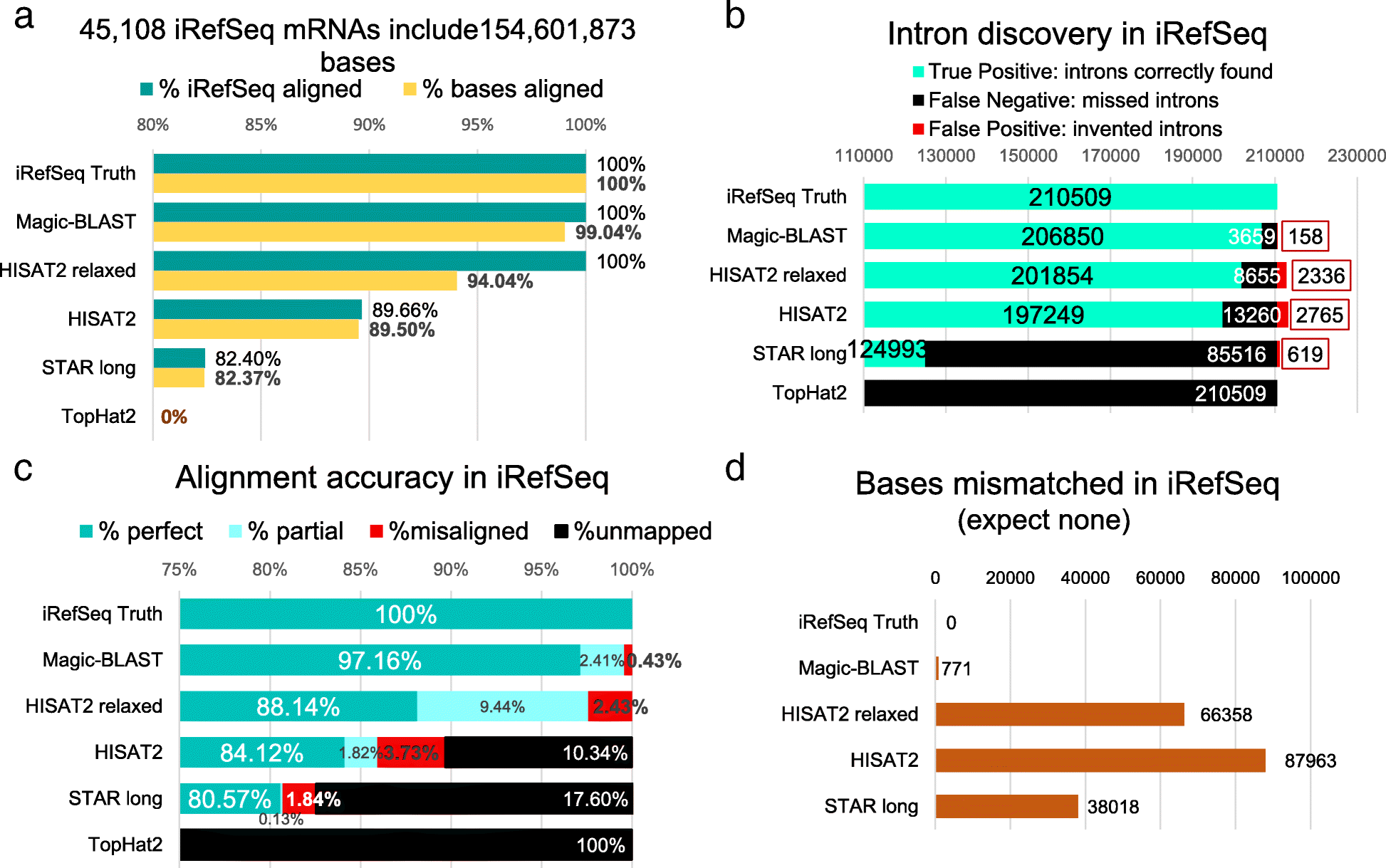 |
|---|
| © Grzegorz M. Boratyn, et al. |
Short reads aligner compartment







