TCGA Database with R
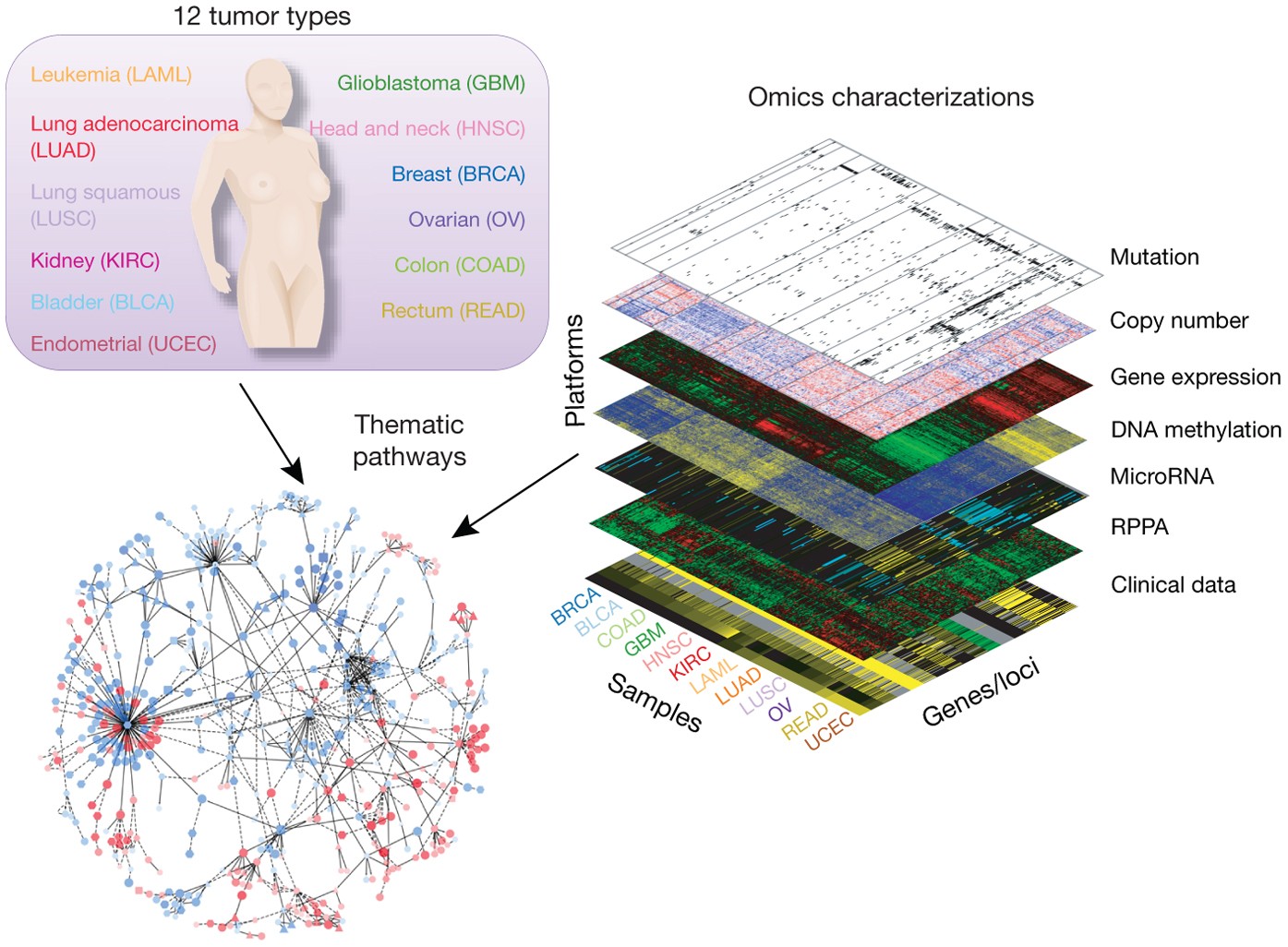
TCGA Database
Reference:
|
If the query is correct, you would see the red codes below and you could start to download the data now.
--------------------------------------
o GDCquery: Searching in GDC database
--------------------------------------
Genome of reference: hg38
--------------------------------------------
oo Accessing GDC. This might take a while...
--------------------------------------------
ooo Project: TCGA-ESCA
ooo Project: TCGA-SARC
ooo Project: TCGA-CESC
ooo Project: TCGA-UCEC
--------------------
oo Filtering results
--------------------
ooo By data.type
ooo By workflow.type
----------------
oo Checking data
----------------
ooo Checking if there are duplicated cases
ooo Checking if there are results for the query
-------------------
o Preparing output
-------------------
Check the group and counts information
|
TCGA-DX-A6Z0-01A-13R-A36F-07 TCGA-X2-A95T-01A-11R-A37L-07 TCGA-DX-A6BF-01A-11R-A30C-07 TCGA-DX-A1L1-01A-11R-A24X-07 ENSG00000000003.15 3415 861 316 4004 ENSG00000000005.6 340 4 14 0 ENSG00000000419.13 2296 905 938 3935 ENSG00000000457.14 594 454 85 595 ENSG00000000460.17 626 318 62 458 ENSG00000000938.13 259 138 271 381
why download data
Sometimes, you may receive errors:
Error in GDCquery(project = projects[3], data.category = "Transcriptome Profiling", : Please set a valid workflow.type argument from the list below: => STAR - Counts
You can’t turn the “GDCprepare” results into data directly. You need to download it first and convert it by “GDCprepare”. See details in github
Differential Expression Genes
Reference: rdrr.io
I am failed to get the expression matrix by using GDCprepare. According to [© g27182818, 2022], it caused by STAR-Count files has more infor than GDCprepare need. What ever, a modified solution could be like codes below:
|
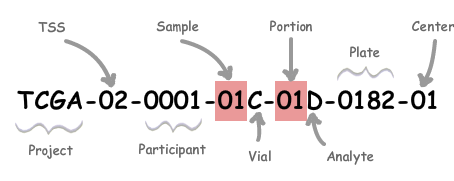
The meaning of the barcode
 |
|---|
| © NIH, GDC |
| Label | Identifier for | Value | Value Description | Possible Values |
|---|---|---|---|---|
| Analyte | Molecular type of analyte for analysis | D | The analyte is a DNA sample | See Code Tables Report |
| Plate | Order of plate in a sequence of 96-well plates | 182 | The 182nd plate | 4-digit alphanumeric value |
| Portion | Order of portion in a sequence of 100 - 120 mg sample portions | 1 | The first portion of the sample | 01-99 |
| Vial | Order of sample in a sequence of samples | C | The third vial | A to Z |
| Project | Project name | TCGA | TCGA project | TCGA |
| Sample | Sample type | 1 | A solid tumor | Tumor types range from 01 - 09, normal types from 10 - 19 and control samples from 20 - 29. See Code Tables Report for a complete list of sample codes |
| Center | Sequencing or characterization center that will receive the aliquot for analysis | 1 | The Broad InstituteGCC | See Code Tables Report |
| Participant | Study participant | 1 | The first participant from MD Anderson for GBM study | Any alpha-numeric value |
| TSS | Tissue source site | 2 | GBM (brain tumor) sample from MD Anderson | See Code Tables Report |
So, the most important information for us is the sample type: Tumor types range from 01 - 09, normal types from 10 - 19 and control samples from 20 - 29. See Code Tables Report for a complete list of sample codes
Abbreviations of projects
| Study Abbreviation | Study Name |
|---|---|
| LAML | Acute Myeloid Leukemia |
| ACC | Adrenocortical carcinoma |
| BLCA | Bladder Urothelial Carcinoma |
| LGG | Brain Lower Grade Glioma |
| BRCA | Breast invasive carcinoma |
| CESC | Cervical squamous cell carcinoma and endocervical adenocarcinoma |
| CHOL | Cholangiocarcinoma |
| LCML | Chronic Myelogenous Leukemia |
| COAD | Colon adenocarcinoma |
| CNTL | Controls |
| ESCA | Esophageal carcinoma |
| FPPP | FFPE Pilot Phase II |
| GBM | Glioblastoma multiforme |
| HNSC | Head and Neck squamous cell carcinoma |
| KICH | Kidney Chromophobe |
| KIRC | Kidney renal clear cell carcinoma |
| KIRP | Kidney renal papillary cell carcinoma |
| LIHC | Liver hepatocellular carcinoma |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| DLBC | Lymphoid Neoplasm Diffuse Large B-cell Lymphoma |
| MESO | Mesothelioma |
| MISC | Miscellaneous |
| OV | Ovarian serous cystadenocarcinoma |
| PAAD | Pancreatic adenocarcinoma |
| PCPG | Pheochromocytoma and Paraganglioma |
| PRAD | Prostate adenocarcinoma |
| READ | Rectum adenocarcinoma |
| SARC | Sarcoma |
| SKCM | Skin Cutaneous Melanoma |
| STAD | Stomach adenocarcinoma |
| TGCT | Testicular Germ Cell Tumors |
| THYM | Thymoma |
| THCA | Thyroid carcinoma |
| UCS | Uterine Carcinosarcoma |
| UCEC | Uterine Corpus Endometrial Carcinoma |
| UVM | Uveal Melanoma |
TCGA Database with R







