
AI Tools for Protein Structures
AlphaFold2
| MSA is important | |
|---|---|
 |
How does the Multiple Sequence Alignment (MSA) help: the sequences alignment could help to encode the residues contact map. When a residues mutated, the contact residues are likely to be mutated, too. This kind of features could be captured by models. |
ESM2
ESM, or Evolutionary Scale Modeling, is a family of protein language models developed by Meta AI. ESM2 is the second generation of this model, which has been trained on a larger dataset and incorporates several architectural improvements over its predecessor, ESM-1b.
How it was trained
ESM-2 is trained to predict the identity of amino acids that have been randomly masked out of protein sequences. This is similar to how models like BERT are trained for natural language processing tasks. The model learns to understand the context of a protein sequence by predicting the masked amino acids based on the surrounding sequence.
Transformer models that are trained with masked language modeling are known to develop attention patterns that correspond to the residue-residue contact map of the protein
ESMFold is a fully end-to-end single-sequence structure predictor, by training a folding head for ESM-2
 |
|---|
 |
| © Zeming Lin |
trRosetta
transform-restrained Rosetta
They inverted this network to generate new protein sequences from scratch, aiming to design proteins with structures and functions not found in nature.By conducting Monte Carlo sampling in sequence space and optimizing the predicted structural features, they managed to produce a variety of new protein sequences.
RFdiffusion
 |
Watson, Joseph L., et al[1] published the RFdiffusion at github in 2023. It fine-tune the RoseTTAFold[2] and designed for tasks like: protein monomer design, protein binder design, symmetric oligomer design, enzyme active site scaffolding and symmetric motif scaffolding for therapeutic and metal-binding protein design. It is a very powerful tool according to the paper. It is based on the Denoising diffusion probabilistic models (DDPMs) which is a powerful class of machine learning models demonstrated to generate new photorealistic images in response to text prompts[3]. |
They use the ProteinMPNN[4] network to subsequently design sequences encoding theses structure. The diffusion model is based on the DDPMs. It can not only design a protein from generation, but also able to predict multiple types of interactions as is shown of the left. It was based on the RoseTTAFold.
Compared with AF2
- AlphaFold2 is like a very smart detective that can figure out the 3D shape of a protein just by looking at its amino acid sequence. On the other hand, RFdiffusion is more like an architect that designs entirely new proteins with specific properties. Instead of just figuring out shapes, it creates new proteins that can do things like bind to specific molecules or perform certain reactions. This makes it incredibly useful for designing new therapies and industrial enzymes.
ImmuneBuilder
ImmuneBuilder: Deep-Learning models for predicting the structures of immune proteins
Method of ABodyBuilder2
- Residues encoding: The sequences are encoded using a one-hot encoding scheme which make the model more efficient and way faster than other methods. The orientation of the residues are also encoded.
- There are 8 block to update the encoding features sequencially. Unly like the AF, the weight are difference and not shared.
- Training data set: 7084 structures from SAbDab. Filtering: No missing residues and resolution ≤ 3.5 Å
- Architect: The architecture of the deep learning model behind ABodyBuilder2 is an antibody-specific version of the structure module in AlphaFold-Multimer with several modifications
- Frame Aligned Point Error (FAPE) loss (like AFM)
- Model selection: Unlike the AF2, ABodyBuilder2 doesn’t rank and select the best structure. It calculate the similarities and select the one most closing to the everage structure. They mentioned that it could “reduces the method’s sensitivity to small fluctuations in the training set. It also results in a small improvement in prediction accuracy.”
A set of deep learning models trained to accurately predict the structure of antibodies (ABodyBuilder2), nanobodies (NanoBodyBuilder2) and T-Cell receptors (TCRBuilder2). ImmuneBuilder generates structures with state of the art accuracy while being much faster than AlphaFold2.
Experience it online: Google Colab
GitHub: oxpig/ImmuneBuilder
They have built three models
- ABodyBuilder2, an antibody-specific model
- NanoBodyBuilder2, a nanobody-specific model
- TCRBuilder2, a TCR-specific model.
How: compare 34 antibody structures recently added
Method | CDR-H1 | CDR-H2 | CDR-H3 | Fw-H | CDR-L1 | CDR-L2 | CDR-L3 | Fw-L |
|---|---|---|---|---|---|---|---|---|
ABodyBuilder (ABB) | 1.53 | 1.09 | 3.46 | 0.65 | 0.71 | 0.55 | 1.18 | 0.59 |
ABlooper (ABL) | 1.18 | 0.96 | 3.34 | 0.63 | 0.78 | 0.63 | 1.08 | 0.61 |
IgFold (IgF) | 0.86 | 0.77 | 3.28 | 0.58 | 0.55 | 0.43 | 1.12 | 0.60 |
EquiFold (EqF) | 0.86 | 0.80 | 3.29 | 0.56 | 0.47 | 0.41 | 0.93 | 0.54 |
AlphaFold-M (AFM) | 0.86 | 0.68 | 2.90 | 0.55 | 0.47 | 0.40 | 0.83 | 0.54 |
ABodyBuilder2 (AB2) | 0.85 | 0.78 | 2.81 | 0.54 | 0.46 | 0.44 | 0.87 | 0.57 |
What is an acceptable RMSD?
The experimental error in protein structures generated via X-ray crystallography has been estimated to be around 0.6Å for regions with organised secondary structures (such as the antibody frameworks) and around 1Å for protein loops.[^Eyal_E]
Side Chain Prediction
ABlooper and IgFold only predict the position of backbones, leaving the side chain to OpenMM[5] and Rosetta[6], while EquiFold, AlphaFold-Multimer and ABodyBuilder2, all of which output all-atom structures.
ABodyBuilder3

Like the ABodyBuilder2, the ABodyBuilder3 is comming from the Deane Lab at University of Oxford[7]. They updated this model in 2024 by bring some new features and slightly improved the performance.
Upgrading:
- In this time, they removed super-long CDRH3 (>=30) antibodies from Bovine.
- OpenMM[5:1] and YASARA[8] were test for structure refinement. They found that YASARA is better. (By checking the result from Supplemental, the results are pretty much the same)
- Language Embedding. In ABodyBuilder3, they now given the options of LM embedding the sequences. As it’s show under the table below, the performance is slightly improved in CDRH3 region. They used the ProtT5[9] model and embedding the H chain and L chain separately. They also tried paired IgT5 and IgBert[10] from the same lab and found the general model is better.
| CDRH1 | CDRH2 | CDRH3 | Fw-H | CDRL1 | CDRL2 | CDRL3 | Fw-L | |
|---|---|---|---|---|---|---|---|---|
| ABodyBuilder2 | 0.41 | 0.38 | 0.57 | 0.50 | 0.47 | 0.48 | 0.72 | 0.40 |
| ABodyBuilder3 | 0.58 | 0.26 | 0.61 | 0.48 | 0.60 | 0.20 | 0.68 | 0.67 |
| ABodyBuilder3-LM | 0.69 | 0.36 | 0.73 | 0.39 | 0.72 | 0.52 | 0.68 | 0.58 |
Equifold
Designing proteins to achieve specific functions often requires in silico modeling of their properties at high throughput scale and can significantly benefit from fast and accurate protein structure prediction. We introduce EquiFold, a new end-to-end differentiable, SE(3)-equivariant, all-atom protein structure prediction model. EquiFold uses a novel coarse-grained representation of protein structures that does not require multiple sequence alignments or protein language model embeddings, inputs that are commonly used in other state-of-the-art structure prediction models. Our method relies on geometrical structure representation and is substantially smaller than prior state-of-the-art models. In preliminary studies, EquiFold achieved comparable accuracy to AlphaFold but was orders of magnitude faster. The combination of high speed and accuracy make EquiFold suitable for a number of downstream tasks, including protein property prediction and design.
https://github.com/Genentech/equifold
IgFold
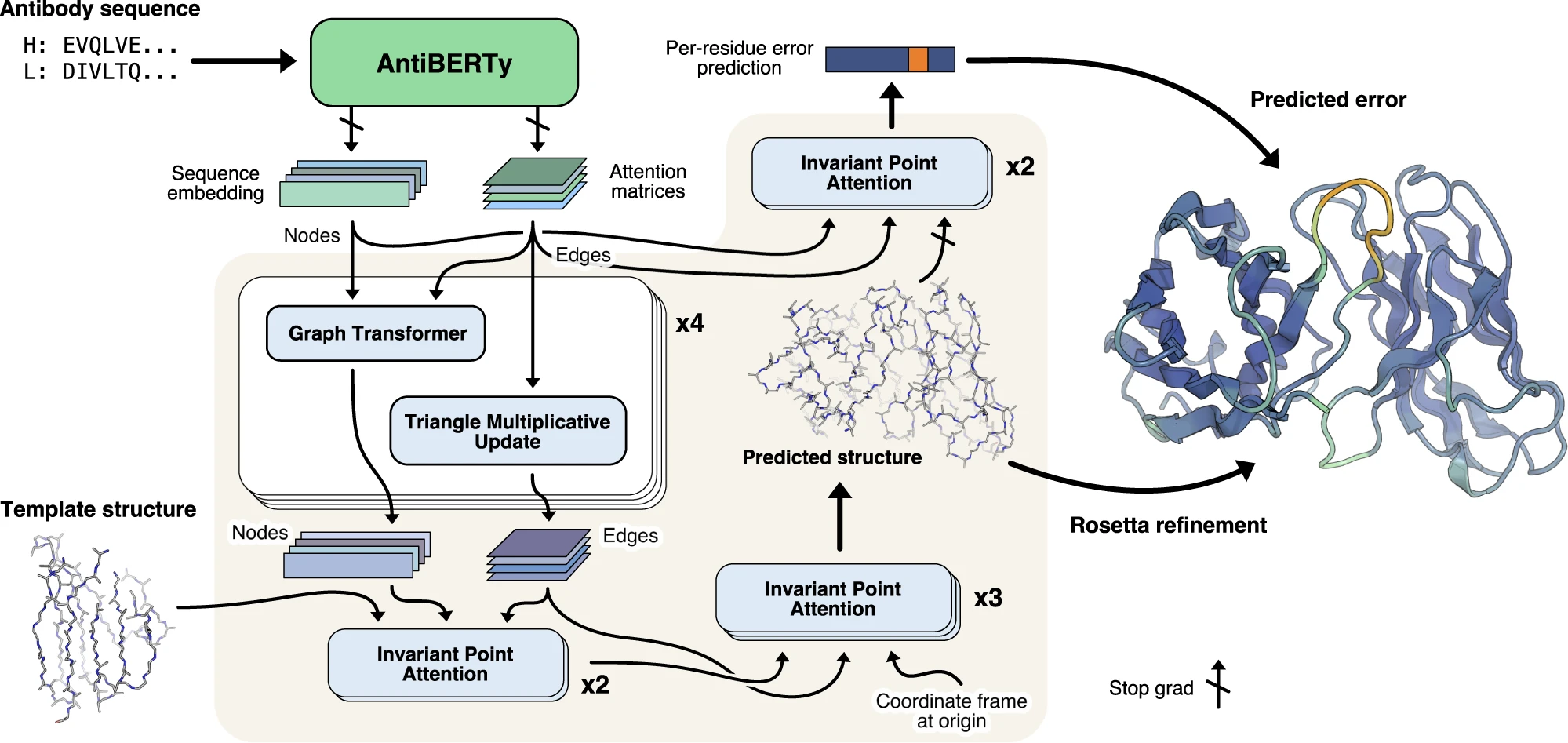
IgFold[11] was developed by the Jeffrey Gray Lab at Johns Hopkins University. It is a fine-tuned AlphaFold-Multimer model. But instead of doing multiple sequences alignments, it embedding the residues like the esm. They trained a large dataset of natural antibodies based on Birt, which is AntiBERTy[12] and trained on 500 million antibody sequences from the SAbDab database. In the folding module, it using full connected residues network, an type of optimized GNN. As you can see the graph from the AntiBERTy below, the LM can do very well on CDRH1 and CDRH2 but not very good on CDRH3, With the help of the AbtiBERTy, it can predict the structures much faster. For the structure prediction training, not only 4K unique antibodies structures from PDB they used, but also about 16K of AF2 predicted structures (pLDDT >=85) are used[13].


 |
|---|
| AntiBERTy was lay in the bioRxiv for many years. Then time when they finally published it, they iterated the model to IgLM[14]. Instead of the mask model, it became like an RNN model |
Why they including AF2 predicted structures in training set?
because an old machine-learning professor told Jeffery that it would be helpful even thought they are not very accurate. And at the end, the performance of the IgFold works slightly better on the AF2. In the end of the Jeffery's talk, they said that they observed that with out adding AF2 predicted data, the performance is not as good as now. (They only took the structure with good confidence, not all data)
Official repository for IgFold: Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies.
The code and pre-trained models from this work are made available for non-commercial use (including at commercial entities) under the terms of the JHU Academic Software License Agreement. For commercial inquiries, please contact Johns Hopkins Tech Ventures at awichma2@jhu.edu.
Try antibody structure prediction in Google Colab: https://github.com/Graylab/IgFold
Note Personal experience
IgFold is much slower than ABodyBuilder2. I think it could because IgFold is using AntiBERTy to embedding the sequences. But the ABodyBuilder2 is using one-hot encoding. I feel that the IgFold is kind of horrible in CDRH3 regions. It predicted CDRH3 loop in an erect conformation incorrectly. It is worse than ABodyBuilder2. It only takes the perfect Fab sequences. Any longer seqeunces would end up as a mass.
Watson J L, Juergens D, Bennett N R, et al. De novo design of protein structure and function with RFdiffusion[J]. Nature, 2023, 620(7976): 1089-1100. ↩︎
Baek M, et al. Accurate prediction of protein structures and interactions using a 3-track network. Science. July 2021. ↩︎
Ramesh, A. et al. Zero-shot text-to-image generation. in Proc. 38th International Conference on Machine Learning Vol. 139 (eds Meila, M. & Zhang, T.) 8821–8831 (PMLR, 2021). ↩︎
Dauparas J, Anishchenko I, Bennett N, et al. Robust deep learning–based protein sequence design using ProteinMPNN[J]. Science, 2022, 378(6615): 49-56. ↩︎
Eastman, P. et al. OpenMM 7: rapid development of high-performance algorithms for molecular dynamics. PLoS Comput. Biol. 13, e1005659 (2017). ↩︎ ↩︎
Alford, R. F. et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Comput. 13, 3031–3048 (2017). ↩︎
Kenlay H, Dreyer F A, Cutting D, et al. ABodyBuilder3: improved and scalable antibody structure predictions[J]. Bioinformatics, 2024, 40(10): btae576. ↩︎
Krieger E, Vriend G. New ways to boost molecular dynamics simulations. J Comput Chem 2015;36:996–1007. ↩︎
Elnaggar A, Heinzinger M, Dallago C et al. Prottrans: toward understanding the language of life through self-supervised learning. IEEE Trans Pattern Anal Mach Intell 2021;44:7112–27. ↩︎
Kenlay H, Dreyer F A, Kovaltsuk A, et al. Large scale paired antibody language models[J]. PLOS Computational Biology, 2024, 20(12): e1012646. ↩︎
Ruffolo J A, Chu L S, Mahajan S P, et al. Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies[J]. Nature communications, 2023, 14(1): 2389. ↩︎
Ruffolo J A, Gray J J, Sulam J. Deciphering antibody affinity maturation with language models and weakly supervised learning[J]. arXiv preprint arXiv:2112.07782, 2021. ↩︎
Jeffrey Gray: Artificial Intelligence Tools for Antibody Engineering and Protein Docking; 2024; YouTube ↩︎
Shuai R W, Ruffolo J A, Gray J J. IgLM: Infilling language modeling for antibody sequence design[J]. Cell Systems, 2023, 14(11): 979-989. e4. ↩︎
AI Tools for Protein Structures







