
Whole Genome Sequencing (WGS)
Whole Genome Sequencing (WGS)
Whole-genome sequencing (WGS) is a comprehensive method for analyzing entire genomes. Genomic information has been instrumental in identifying inherited disorders, characterizing the mutations that drive cancer progression, and tracking disease outbreaks. Rapidly dropping sequencing costs and the ability to produce large volumes of data with today’s sequencers make whole-genome sequencing a powerful tool for genomics research. (Illumina)
Illumina WGS
KEY WHOLE-GENOME SEQUENCING METHODS
- Large whole-genome sequencing
- Small whole-genome sequencing
- De novo sequencing
- Targeting to species without reference genome
- Phased sequencing
- Human whole-genome sequencing
- optimized for human
- Long-reads sequencing
Examples from Publications
SKLA1.0 (Duck)
| A chromosome-scale Beijing duck assembly (SKLA1.0[1]) by integrating Nanopore, Bionano, and Hi-C data covers 40 chromosomes, improves the contig N50 of the previous duck assembly with highest contiguity (ZJU1.0) of more than a 5.79-fold and contains a complete genomic map of the MHC. |  © Olivia Crowe © Olivia Crowe |
Species: Anas platyrhynchos (a native breed in China, using a hierarchical and hybrid approach)
Reads types: Nanopore, Bionano, and Hi-C data.
- 71-fold normal and 24-fold ultra-long Nanopore reads
- 117-fold 150bp paired-end Illumina reads for polishing
- 216-fold optical map reads and 234-fold PE150 Hi-C
Result:
- 40 chromosomes, improves the contig N50 of the previous duck assembly with highest contiguity (ZJU1.0) of more than a 5.79-fold
- a complete genomic map of the MHC
Solved challenges:
- traditional assembly tools have not enabled proper genomic draft of highly repetitive and GC-rich sequences, such as the MHC
Something I don’t understand:
- C18 Duck?
- heterozygosity estimation: why they do it? How could it help on the genome assembly?
- What is BUSCO score?
Steps for Genome assembly:
- Estimate Genome Heterozygosity
- Before starting the assembly, the genome heterozygosity of the C18 duck was estimated. The heterozygosity was found to be as low as 0.58% (Additional file 1: Table S1 and Additional file 2: Fig. S1-S3).
- Assemble Genome with Nanopore Reads
- Using 71-fold normal and 24-fold ultra-long Nanopore reads, the duck genome was assembled into 151 contigs covering a total length of 1.22 Gb with a contig N50 of 32.81 Mb (Additional file 1: Table S2-S3).
- NextDenovo: Clean and assembly
- Polish Contigs with Illumina Reads
- The 151 contigs were then polished with 912 million 150-bp Illumina pair-end reads, corrected, and integrated with high-quality optical maps (Additional file 1: Table S4-S5). This effort generated 69 scaffolds with a scaffold N50 of 72.53 Mb (Additional file 1: Table S6).
- Nextpolish-1.2.3[2]: polished three rounds
- Use Hi-C Data for Scaffold Ordering
- Perform Gap Filling
- Gap filling was performed using 95-fold corrected Nanopore reads to remove gaps, generating the final duck assembly (SKLA1.0), representing 1.16 Gb of the genomic sequence, approximately 99.11% of the estimated genome size (Table 1).
- Gapcloser-0.56[7]
- Chromosome Coverage and Comparison
- Since the duck contains 80 chromosomes (diploid, 2n=80), it was inferred that this duck assembly had covered all chromosomes except W (Additional file 1: Table S8). The SKLA1.0 assembly was compared with the previous duck BGI_duck_1.0 assembly, the ZJU1.0 assembly, and two high-quality avian reference genomes (chicken GRCg6a and zebra finch bTaeGut1.4.pri). These analyses indicated that the SKLA1.0 assembly represents a major improvement over the previous assemblies in terms of contiguity, completeness, and chromosome size. The contiguity and completeness of SKLA1.0 is also higher than that of the zebra finch bTaeGut1.4.pri and the chicken GRCg6a (Fig. 1a–d and Table 1).
After Assembly
- Funannotate pipeline and the GETA pipeline together with a manual curation of key gene families: 17,896 duck coding genes. Quality was validated by number of coding genes, # of transcripts, # of gaps, and BUSCO score.
- Visualization: Bionano map-SOLVE
ASM2904224v1: Greater scaup (Aythya marila)
| First high-quality chromosome-level genome assembly of A. marila[8], with a final genome size of 1.14 Gb, scaffold N50 of 85.44 Mb, and contig N50 of 32.46 Mb. A total of 154.94 Mb of repetitive sequences were identified. 15,953 protein-coding genes were predicted in the genome, and 98.96% of genes were functionally annotated. | © Olivia Crowe |
- Nanopore long reads, errors corrected using Illumina short reads
- Quality: Final size: 1.14 Gb, scaffold N50 of 85.44 Mb, and contig N50 of 32.46 Mb.
- 106 contigs were clustered and ordered onto 35 chromosomes based on Hi-C data, covering approximately 98.28% of the genome
- BUSCO assessment showed that 97.0% of the highly conserved genes
Source: muscle tissue of wild male.
- 60.77 GB for Illumina HiSeq 4000: Illumina® TruSeq® Nano DNA Library Prep kits to generate sequencing libraries of genomic DNA
- 122.55 GB from PromethION platform (91.36 fold of the greater scaup’s genome)
- 63.43 Gb for Hi-C data
Genome Assembly
- Quality Control:
- K = 17, the estimated genome size was 1,341.4 Mb, the heterozygosity was 0.47%, and the proportion of repetitive sequences was 42.28%
- jellyfish (v2.2.7)
- Assembly:
- assemble the genome with Oxford nanopore technologies (ONT) long reads
- NextDenovo (v2.4.0)
- Polish:
- increase the precision of single base with Illumina short reads
- NextPolish12 (v1.3.1)[2:1]
- Scaffold Ordering:
- mount the contigs in preliminarily assembly onto chromosomes based on the signal strength after Hi-C data
- ALLHiC (v0.9.8)[9] and Juicebox (v1.11.08)
| HiC Results for the global heat map of all the chromosomes. |
|---|
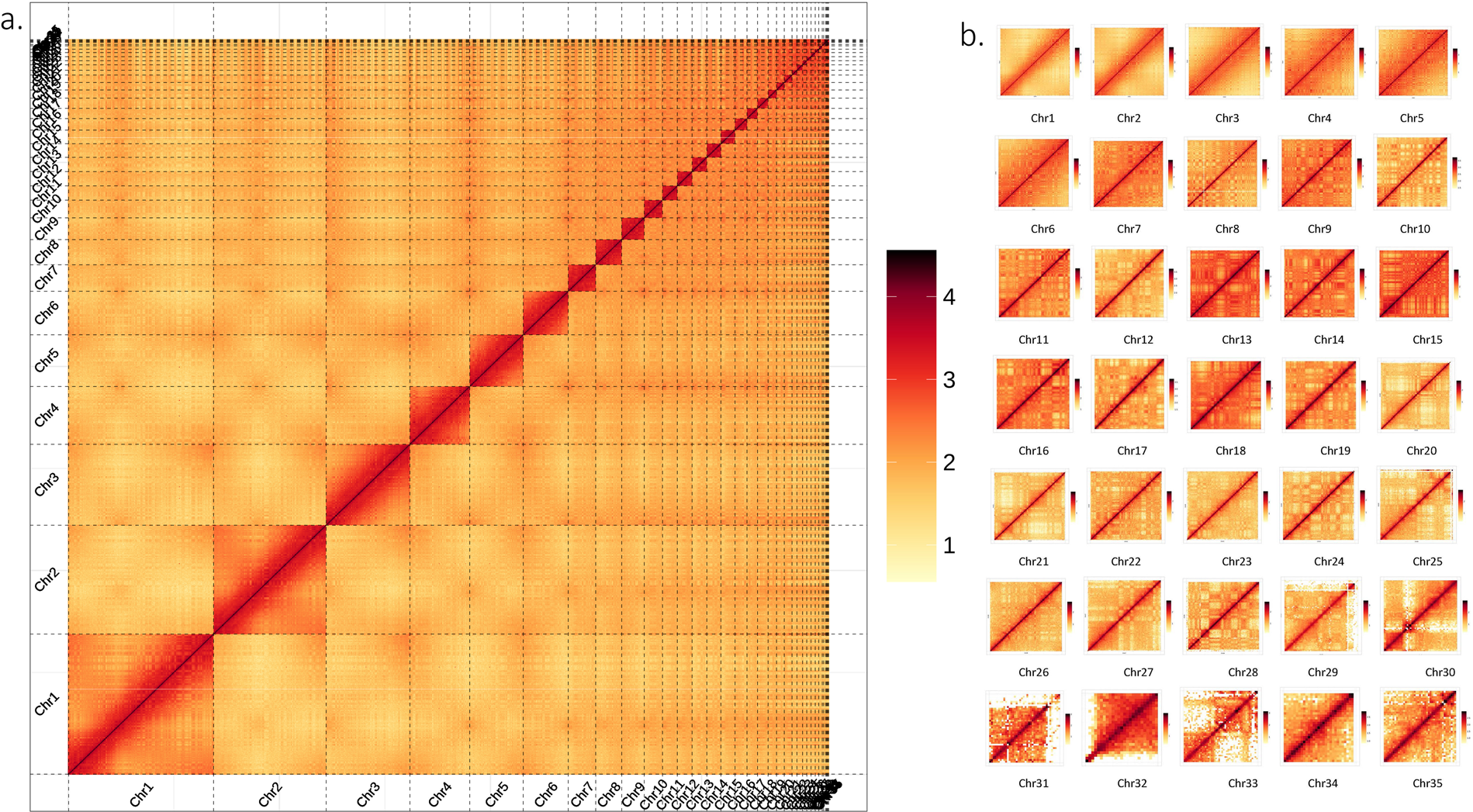 |
| © Shengyang Zhou |
After Assembly
- Assessment
- Burrows-Wheeler aligner14 (BWA) (v0.7.8) to map Illumina reads to the genome with matching rate was approximately 98.80%.
- Merqury15 (v1.3) was ran to evaluate assembly quality value (QV), and a high QV (42.14)
- Benchmarking Universal Single-Copy Orthologs16 (BUSCO) (v5.4.4) (use option “–augustus”) and Core Eukaryotic Genes Mapping Approach17 (CEGMA) (v2.5) were also used to assess the integrity
- 238 of 248 core eukaryotic genes were detected using CEGMA
- Comparison
- Mummer18 (v4.0.0) was used to identify the synteny between A. marila and tufted duck19 (Aythya fuligula) genomes to determine orthologous chromosome pairs, and we used TBtools20 (v1.112) to draw the synteny between their chromosomes.
- Annotation of repetitive sequences
- de novo prediction: Tandem Repeats Finder21 (TRF) (v4.09) to detect tandem repeat sequences
- RepeatModeler (v2.0.3), RepeatScout22 (v1.0.6) and RECON (v1.08) were used to build a database of transposable element (TE)
- RepeatProteinMask and RepeatMasker (v4.1.2-p1) were used for homology prediction with Repbase database23 and Dfam database24, the species parameter used was chicken.
- Gene structure prediction
- Prediction Methods:
- Ab Initio Prediction: Used software Augustus (v3.3.2), GlimmerHMM (v3.0.4), and Geneid (v1.4.5).
- Homology-Based Prediction: Utilized genomes and annotation files from six related species (Anser cygnoides, Anas platyrhynchos, Aythya fuligula, Cygnus olor, Cygnus atratus, Gallus gallus) downloaded from NCBI.
- RNA-Seq Prediction: Processed raw data from six transcriptomic samples using fastp (v0.23.1), assembled paired-end reads with SPAdes (v3.15.3), identified candidate coding regions using TransDecoder (v5.5.0), and clustered sequences using CD-hit (v4.8.1).
- Integration:
- Matching and Splicing: Protein sequences from related species were matched to the A. marila genome using Spaln (v2.4.6) and accurately spliced with GeneWise (v2.4.1).
- Gene Set Generation: Combined homology-based, RNA-Seq, and ab initio predictions using EvidenceModeler (EVM) (v1.1.1) and incorporated masked repeats.
- Prediction Methods:
- Databases and Tools:
- DIAMOND: Used for sequence alignment against SwissProt, TrEMBL, NR (Non-Redundant Protein Sequence Database), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes Orthology (KO) databases, with an e-value cutoff of 1e-5.
- InterPro: Utilized for classifying proteins into families and predicting domains and important sites using InterProScan (v5.53).
- Filtering and Verification of Gene Set for A. marila
- Ortholog Identification:
- OrthoFinder: Used to identify orthologs among A. marila and six related species.
- Resulted in 4,086 unassigned genes, of which 3,421 were not annotated in any database.
- Filtering Process:
- Most unannotated genes (3,417/3,421) were predicted by at least one de novo prediction software, with only four supported by other evidence.
- Removal of these unassigned genes did not affect the BUSCO test results, indicating they may not represent real genes.
- Final Gene Set:
- After filtering out unassigned genes without annotations and 159 prematurely terminated genes, 15,953 genes remained, including 182 partial genes.
- 98.96% of the final gene set was annotated.
- Ortholog Identification:
Pig Sscrofa11.1
Warr A, Affara N, Aken B, et al. An improved pig reference genome sequence to enable pig genetics and genomics research[J]. Gigascience, 2020, 9(6): giaa051.
TJ Tabasco
corrected and assembled using Falcon (v.0.4.0)
65-fold coverage (176 Gb) of the genome
3,206 contigs with a contig N50 of 14.5 Mb.
Compare
contigs were mapped to the previous draft assembly (Sscrofa10.2) using Nucmer
gap closure using PBJelly
| Statistic | Sscrofa10.2 | Sscrofa11 | Sscrofa11.1 | USMARCv1.0 | GRCh38.p13 |
|---|---|---|---|---|---|
| Total sequence length | 2,808,525,991 | 2,456,768,445 | 2,501,912,388 | 2,755,438,182 | 3,099,706,404 |
| Total ungapped length | 2,519,152,092 | 2,454,899,091 | 2,472,047,747 | 2,623,130,238 | 2,948,583,725 |
| No. of scaffolds | 9,906 | 626 | 706 | 14,157 | 472 |
| Gaps between scaffolds | 5,323 | 24 | 93 | 0 | 349 |
| No. of unplaced scaffolds | 4,562 | 583 | 583 | 14,136 | 126 |
| Scaffold N50 | 576,008 | 88,231,837 | 88,231,837 | 131,458,098 | 67,794,873 |
| Scaffold L50 | 1,303 | 9 | 9 | 9 | 16 |
| No. of unspanned gaps | 5,323 | 24 | 93 | 0 | 349 |
| No. of spanned gaps | 233,116 | 79 | 413 | 661 | 526 |
| No. of contigs | 243,021 | 705 | 1,118 | 14,818 | 998 |
| Contig N50 | 69,503 | 48,231,277 | 48,231,277 | 6,372,407 | 57,879,411 |
| Contig L50 | 8,632 | 15 | 15 | 104 | 18 |
| No. of chromosomes* | *21 | 19 | *21 | *21 | 24 |
pig (Sscrofa10.2, Sscrofa11.1, USMARCv1.0), human (GRCh38.p13)
Hu J, Song L, Ning M, Niu X, Han M, Gao C, Feng X, Cai H, Li T, Li F, Li H, Gong D, Song W, Liu L, Pu J, Liu J, Smith J, Sun H, Huang Y. A new chromosome-scale duck genome shows a major histocompatibility complex with several expanded multigene families. BMC Biol. 2024 Feb 5;22(1):31. doi: 10.1186/s12915-024-01817-0. PMID: 38317190; PMCID: PMC10845735. Paper ↩︎
Hu J, Fan JP, Sun ZY, Liu SL. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 2020;36:2253–5. ↩︎ ↩︎
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. ↩︎
Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016;3:95–8. ↩︎
Dudchenko O, Batra SS, Omer AD, Nyquist SK, Hoeger M, Durand NC, et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 2017;356:92–5. ↩︎
Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, et al. Juicebox Provides a Visualization System for Hi-C Contact Maps with Unlimited Zoom. Cell Syst. 2016;3:99–101. ↩︎
Xu MY, Guo LD, Gu SQ, Wang O, Zhang R, Peters BA, et al. TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. Gigascience. 2020;9:giaa094–104. ↩︎
Zhou S, Xia T, Gao X, et al. A high-quality chromosomal-level genome assembly of Greater Scaup (Aythya marila)[J]. Scientific Data, 2023, 10(1): 254. ↩︎
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nature Plants 5, 833–845 (2019). ↩︎
Whole Genome Sequencing (WGS)
https://karobben.github.io/2024/06/17/Bioinfor/wholegenomesequencing/







