AlphaFold
AlphaFold2
Main source from:
- YouTube:
- Kendrew Lecture 2021 pt2 - John Jumper: first author of AlphaFold2
- Review and discussion of AlphaFold3; 2024: Sergey Ovchinnikov, MIT
- MRC Laboratory of Molecular Biology; 2024
- What Is AlphaFold? | NEJM
- how AlphaFold actually works
- Papers:
- Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold[J]. nature, 2021, 596(7873): 583-589.
- Abramson J, Adler J, Dunger J, et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3[J]. Nature, 2024, 630(8016): 493-500.

Main architecture of the AlphaFold2:
- Input
- MSA: Multiple Sequence Alignment, to align the sequences to gain the evolutionary information. The idea is: When residues pair-mutated, they are highly likely to be contact with each other. So, the model could learn the contact map from the MSA.
- Pairing: It might the most important part for the model. Captures pairwise relationships between residues.
- At the beginning, there are almost no information.
- triangle inequality: ij+kj>=ik (i, j, k are the residues)
- Structure Database: The model will use the structure database to get the information of the protein structure.
- MSA results would be used to generate the MSA representation. Pairing and Structure Database would be used to generate the Pair representation.
- Evoformer
- The Evoformer is a transformer model which is used to learn the MSA representation and Pair representation. The Evoformer will use the MSA representation and Pair representation to refine the MSA representation and Pair representation
- Structure
- The structure module would use the representations to do rotation and translation to generate the final structure.
- Recycle:
- The final structure would be collected and used to refine the refined representations in the Evoformer module and Structure module multiple times.
Multiple Sequence Alignment (MSA)
Multiple sequences are critical for the prediction of protein structures. The more sequences you have, the better the prediction. Even taking away the sequences in PDB databse, AF2 cans till give a very good prediction. “The sequence is very very clear for the structure” (John)
| MSA is important | |
|---|---|
 |
How does the Multiple Sequence Alignment (MSA) help: the sequences alignment could help to encode the residues contact map. When a residues mutated, the contact residues are likely to be mutated, too. This kind of features could be captured by models. |
 |
Without the MSA, the predicted result is terrible. |
| MAS | pLDDT | PAE |
|---|---|---|
 |
 |
 |
As you can see from this graphic, the depth of the MSA results are difference. The depth would effect the final quality of the model. And the depth of the MSA is also highly positive associated with the pLDDT score. The more sequences you have, the better the prediction. Except the depth, the pLDDT score is also highly related to the second structures. Random loop usually get a low pLDDT.
PAE’s concept is pretty similar to pLDDT. pLDDT is predicts the single residue confidence, while PAE is predicts the pairwise residue confidence. The higher the PAE score, the more confident the model is about the distance between two residues.
Results
- pLDDT: a per-atom confidence estimate on a 0-100 scale where a higher value indicates higher confidence.
- PAE (predicted aligned error): estimate of the error in the relative position and orientation between two tokens in the predicted structure.
- pTM and ipTM scores: the predicted template modeling (pTM) score and the interface predicted template modeling (ipTM) score are both derived from a measure called the template modeling ™ score. This measures the accuracy of the
entire structure
In alphafold2, you’ll get 5 results for each seed because there are 5 different models which trained slightly different. The model would rank the results by pLDDT, pTM, and, ipTM.
You may tell the false positive based on the PEA score
 |
False Negative: The results from the top is predicted incorrectly. The PAE score between chain with different color is very high. The results from the bottom is predicted correctly. The PAE score between chain with different color is very low which means the result my reliable. |
 |
False Positive: Though the PAE are very good, but they are not supposed to be interact to each other. It could be caused by some protein from other families may interacted in this way. So, the model learned this possibilities. |
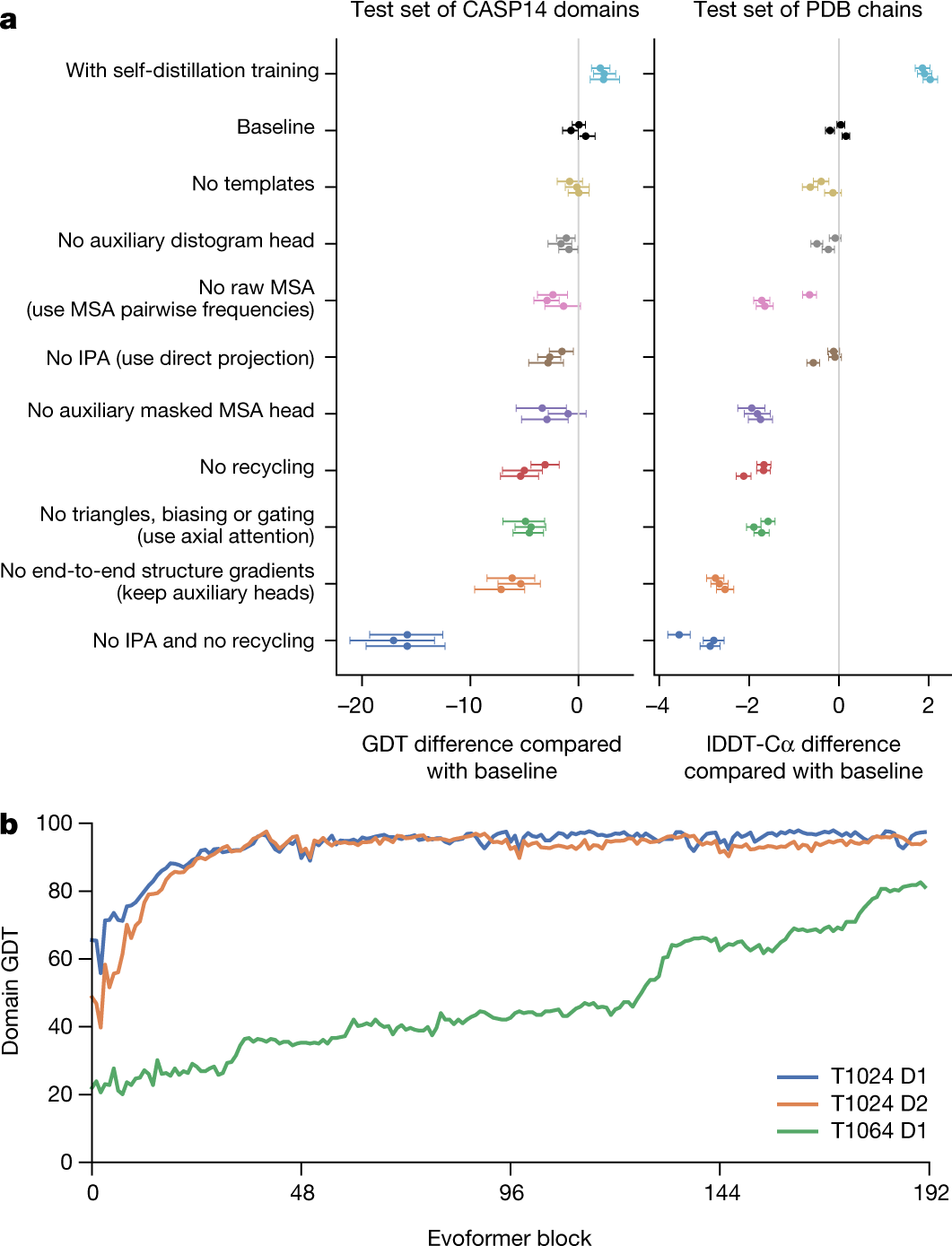 |
|---|
| Ablation results on two target sets: the CASP14 set of domains (n = 87 protein domains) and the PDB test set of chains with template coverage of ≤30% at 30% identity (n = 2,261 protein chains). Domains are scored with GDT and chains are scored with lDDT-Cα. The ablations are reported as a difference compared with the average of the three baseline seeds. Means (points) and 95% bootstrap percentile intervals (error bars) are computed using bootstrap estimates of 10,000 samples. b, Domain GDT trajectory over 4 recycling iterations and 48 Evoformer blocks on CASP14 targets LmrP (T1024) and Orf8 (T1064) where D1 and D2 refer to the individual domains as defined by the CASP assessment. Both T1024 domains obtain the correct structure early in the network, whereas the structure of T1064 changes multiple times and requires nearly the full depth of the network to reach the final structure. Note, 48 Evoformer blocks comprise one recycling iteration. |
Limitations
As you can expect, MSA brings huge convenience to the model. But it also brings the major limitation: it not sensitive to the mutations. In some cases, a point mutation would make the inner structure or interaction between proteins fail. But the MSA would not be able to capture this kind of information and database lacking such structures. So, the model would have high false positive rate. This features makes antibody-antigen interaction prediction very hard. (It was said that RoseTTAFold can do much better on handling mutations.)
On the other hand, as long as AF relies on the MSA, it is not good at fast mutated proteins interactions. For example, the virus protein and antibodies. Virus mutation every years and antibodies mutated every few days[1]. MSA would more reliable on the co-evolution over thousands or millions of years. But for the fast mutated proteins, the MSA would not be able to capture the co-evolution information. So, the model would not be able to learn the structure from scratch.
 |
It is also not hard to imagine that the model would not be able to predict the structure of the protein which has no homologous sequences. The model is not able to learn the structure from scratch. |
 |
It seems like AF2 can get a much better DockQ score then the other models. But the this kind of compairation is very tricky |
John Jumper claimed that the AF2-multimer “performs poorly on antibody interactions”, could be caused by “still miss quite a few interactions”
AlphaFold3
 |
|---|
In AF3, John Jumper became the first corresponding author with Demis. The first author is Josh Abramson.
The main difference between AF2 and AF3 is that AF3 moved the structure model out of the cycling and only put the Pairformer in the cycling. The structure model is only used once. And it is replaced by the Diffusion model. Meanwhile, the contribution of MSA was changed. In Af3, there is an independent MSA module and only 4 blocks. If Af2, MSA would used for 48 blocks. This change makes the training much faster. For 5000 tokens, AF2 would takes about 2 to 3 days. But now, it only takes about 20 minutes.
 |
AF3 made some improvements on monomers and prtotein-protein interaction prediction caompare to AF2. It seems like the AF3 made huge improvement on the protein-antibody prediction. But according to Sergey, this comparision is very tricky. They didn’t take the results by single try. They actually test different seeds independently to get over 1000 results. And then, they took the best result. So, the results are not very reliable. This hind may tell use that diffusion model may not solve the sampling problem well |
- reproducibility: For AF model, if you change the random seeds, the result would be totally different. So, when we try to use it to predict the protein-antibody, we would got all different results and it is hard to tell which one is the true positive.
Why Diffusion model doesn't work well in protein-antibody prediction?
Personally, I think the main limitation still bring from the MSA. The homology modeling method would be ok for protein-protein prediction because this type of interaction could be described and protein family-family interaction. But for antibodies, the interaction is very specific and restricted to the unique loop region which mostly not exist in the any of database. Meanwhile, the contribution of the residues are extremely unbalanced. Some residues credit as motif plays the most important role in the recognition. Rest of residues are maybe play the role in supporting the structure, complementary to the surface to present the motif in a correct position, etc. So, antibodies are more sensitive to the surface structure. But the diffusion model would fail in the initial interface prediction. And during the de-noise process, this error would be amplified. Meanwhile, the model doesn't know if the antibody can dock to the target or not. During the diffusion steps, it only tries is best to dock on it during the de-noise steps and make false positive result.
Running AlphaFold 3 locally
Installing AF3 on a local machine (Conda)
You can install and run AlphaFold 3 locally using a Conda-based setup without system-wide packages. A step-by-step guide:
- Requirements: Linux, NVIDIA GPU (Ampere or newer, e.g. RTX 3060–4090, A100), CUDA drivers, ~700 GB disk (databases ~627 GB, models ~1.1 GB, conda env ~6.7 GB).
- Steps (summary; follow the full guide for exact commands):
- Install Miniconda and create a Python 3.11 conda environment (e.g.
Alphafold3). - Install dev tools and deps via conda (cmake, gcc, Boost, HMMER, zlib, etc.).
- Install Python packages with pip (JAX/CUDA 12, matching your GPU: Ampere/Ada vs Blackwell).
- Clone google-deepmind/alphafold3, download databases (
fetch_databases.sh), obtain and place model parameters (af3.bin). - Install AF3 from the repo with
pip install --no-deps ., runbuild_datain the condabindir, then test withpython run_alphafold.py --help. - Use an execution script (e.g.
AF3_run.sh) that points to HMMER binaries,db_dir,model_dir, and your input JSON.
- Install Miniconda and create a Python 3.11 conda environment (e.g.
Source (full instructions):
- Model3DBio/AlphaFold3-Conda-Install
- Raw README: README.md
Running AF3 without MSA (data pipeline skipped)
By default, AF3 runs a data pipeline that builds MSAs (e.g. with Jackhmmer), which can be slow. Using only --run_data_pipeline=false is not enough: the featurisation step still expects unpaired MSA and will raise ValueError: Protein chain 1 is missing unpaired MSA.
To run without MSA you must also set specific fields in the input JSON for each protein (see issue #151, solution by @alchemistcai):
- Set
unpairedMsaandpairedMsato""explicitly. - Set
templatesto[]explicitly.
JSON template for a protein chain (no MSA):
|
Full fold_input.json example (single protein, no MSA; use with --run_data_pipeline=false):
|
For multiple chains, repeat the protein object (with unique id) and set unpairedMsa, pairedMsa, and templates for each.
Source:
Test run (local AF3)
| Setup | Input | Time |
|---|---|---|
| Local AF3 (no MSA pipeline) | Monomial HA + scFv antibody | 4.5 min |
| Local AF3 (no MSA pipeline) | Trinomial HA + scFv antibody | 26 min |







