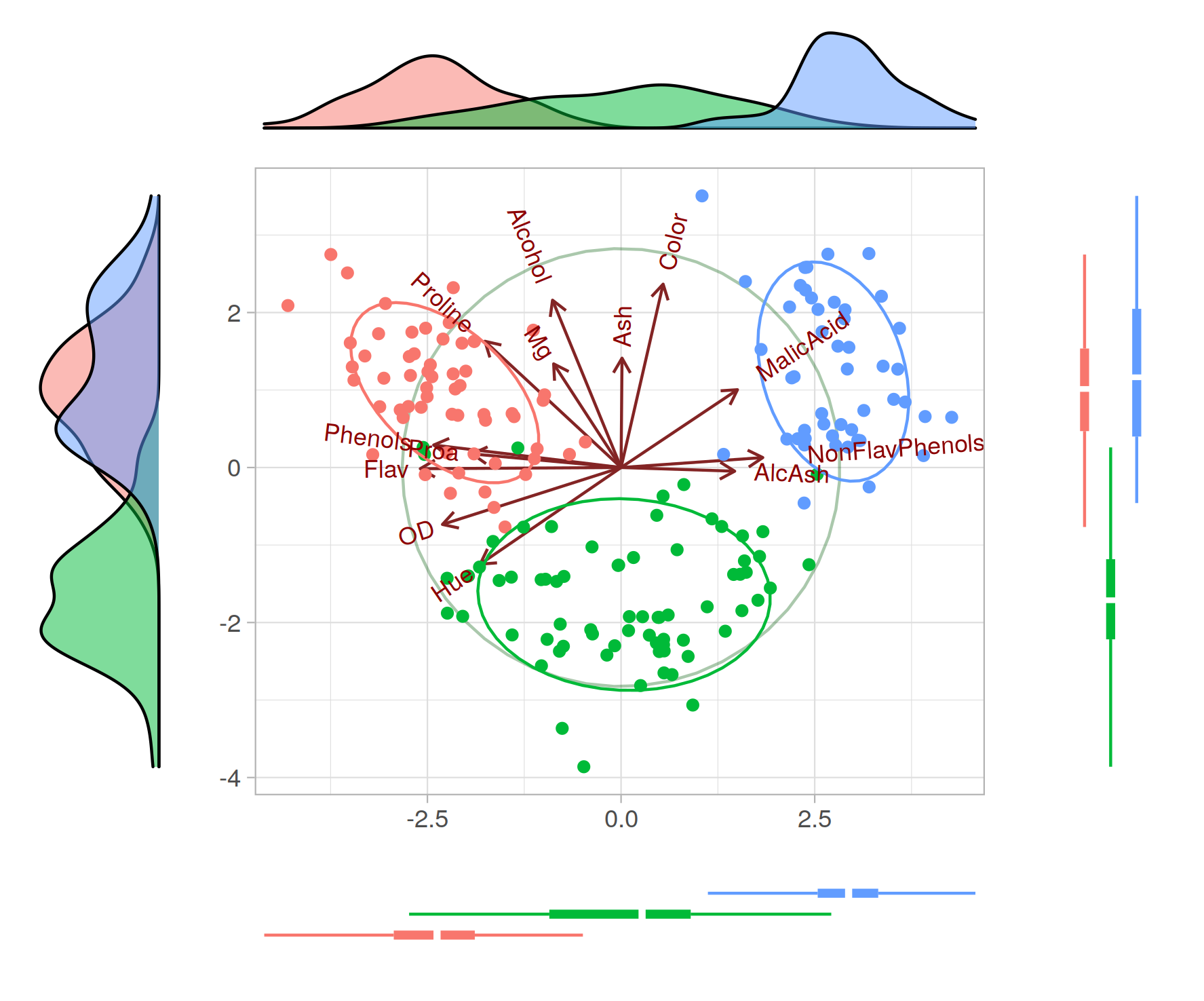
Principal Component Analysis (PCA) and Principal Coordinates Analysis (PCoA, also known as Multidimensional Scaling, MDS) are both techniques used for dimensionality reduction, which is the process of reducing the number of random variables under consideration by obtaining a set of principal variables. However, they are used in different contexts and have different underlying methodologies.
PCA is a technique that is used when you have a multivariate data set and you want to identify new variables that will represent the variability of your entire data set as much as possible. The new variables, or principal components, are linear combinations of the original variables. PCA operates on a covariance (or correlation) matrix, which implies that it is a parametric method.
On the other hand, PCoA is a method used in ecology and biology to transform a matrix of distances (or dissimilarities) between samples into a new set of orthogonal axes, the most important of which can be plotted against each other. PCoA can be applied to any symmetric distance or dissimilarity matrix. Unlike PCA, PCoA is non-parametric and makes no assumptions about the distribution of the original variables.
So, the main difference lies in the type of data they work with: PCA works with the actual data matrix and is used when you have a set of observations and measurements, while PCoA works with a matrix of pairwise distances and is used when you have a set of pairwise dissimilarities (like geographical distances between cities or genetic distances between individuals or species).
import pandas as pd from sklearn.decomposition import PCA from sklearn.preprocessing import StandardScaler from sklearn.datasets import load_wine import matplotlib.pyplot as plt
# Step 2: Standardize the data (optional but recommended) scaler = StandardScaler() scaled_data = scaler.fit_transform(df)
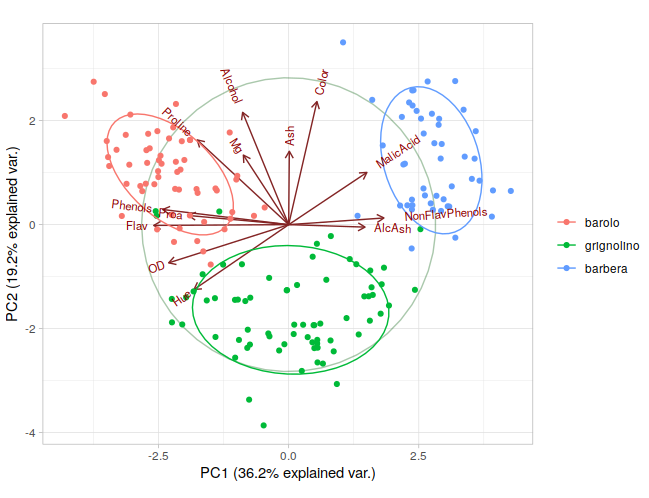
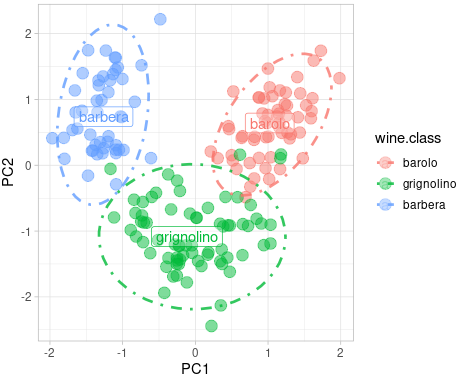
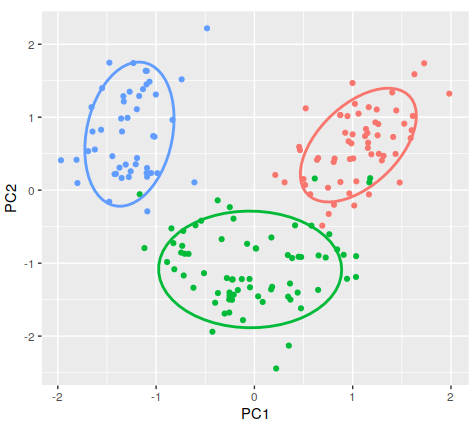
# Run the PCA with 3 components pca = PCA(n_components=2) # Reduce to 2 dimensions for visualization principal_components = pca.fit_transform(scaled_data) pca_df = pd.DataFrame(data=principal_components, columns=['PC1', 'PC2']) pca_df['target'] = wine_data.target # Create a scatter plot with color based on the target class scatter = plt.scatter(pca_df['PC1'], pca_df['PC2'], c=pca_df['target'], cmap='viridis', edgecolor='k', alpha=0.7)
# Adding labels and title plt.xlabel('Principal Component 1') plt.ylabel('Principal Component 2') plt.title('PCA of Wine Dataset')
# Add a color bar to indicate the different classes plt.colorbar(scatter, ticks=[0, 1, 2], label='Wine Class')
# Optionally, add a legend (if your target classes are categorical) # legend = plt.legend(*scatter.legend_elements(), title="Classes") # plt.gca().add_artist(legend)
# Show the plot plt.grid(True) plt.show()
By checking the raw data, each column is feature, row is element.
Index
Alcohol
Malic Acid
…
OD280/OD315 of Diluted Wines
Proline
0
14.23
1.71
…
3.92
1065.0
1
13.20
1.78
…
3.40
1050.0
2
13.16
2.36
…
3.17
1185.0
3
14.37
1.95
…
3.45
1480.0
4
13.24
2.59
…
2.93
735.0
…
…
…
…
…
…
173
13.71
5.65
…
1.74
740.0
174
13.40
3.91
…
1.56
750.0
175
13.27
4.28
…
1.56
835.0
176
13.17
2.59
…
1.62
840.0
177
14.13
4.10
…
1.60
560.0
Explained Variance Ratio
# Step 3: Perform PCA pca = PCA() # By default, PCA will consider all components pca.fit(scaled_data)
# Step 4: Get the explained variance ratio for each component explained_variance_ratio = pca.explained_variance_ratio_
# Step 5: Create a DataFrame to store component contributions component_contributions = pd.DataFrame({ 'Principal Component': [f'PC{i+1}'for i inrange(len(explained_variance_ratio))], 'Explained Variance Ratio': explained_variance_ratio })
# Step 6: Sort the components by their contribution (explained variance ratio) component_contributions_sorted = component_contributions.sort_values(by='Explained Variance Ratio', ascending=False)
# Display the sorted components print(component_contributions_sorted)
# Step 7: Optionally, you can plot the explained variance ratio to visualize plt.figure(figsize=(8, 6)) plt.bar(range(1, len(explained_variance_ratio) + 1), explained_variance_ratio, alpha=0.7, align='center', label='individual explained variance') plt.step(range(1, len(explained_variance_ratio) + 1), explained_variance_ratio.cumsum(), where='mid', label='cumulative explained variance') plt.ylabel('Explained variance ratio') plt.xlabel('Principal components') plt.legend(loc='best') plt.tight_layout() plt.show()
Index
Principal Component
Explained Variance Ratio
0
PC1
0.361988
1
PC2
0.192075
2
PC3
0.111236
3
PC4
0.070690
4
PC5
0.065633
5
PC6
0.049358
6
PC7
0.042387
7
PC8
0.026807
8
PC9
0.022222
9
PC10
0.019300
10
PC11
0.017368
11
PC12
0.012982
12
PC13
0.007952
Contribution of Each Features
import numpy as np
# adding a new column for test. df2 = df.copy() df2['test'] = 0 # Perform PCA pca = PCA() pca.fit(df2) # Get the loadings (components) loadings = pca.components_.T # Calculate the contribution of each feature feature_contributions = np.sum(np.abs(loadings), axis=1) # Create a DataFrame to rank features feature_importance_df = pd.DataFrame({ 'Feature': wine_data.feature_names + ['test'], 'Contribution': feature_contributions }) feature_importance_df = feature_importance_df.sort_values(by='Contribution', ascending=False).reset_index(drop=True) # Rank features by their contribution
We can find the this contribution results, the text column doesn’t has any of contribution in variation since all values are equals to 1.