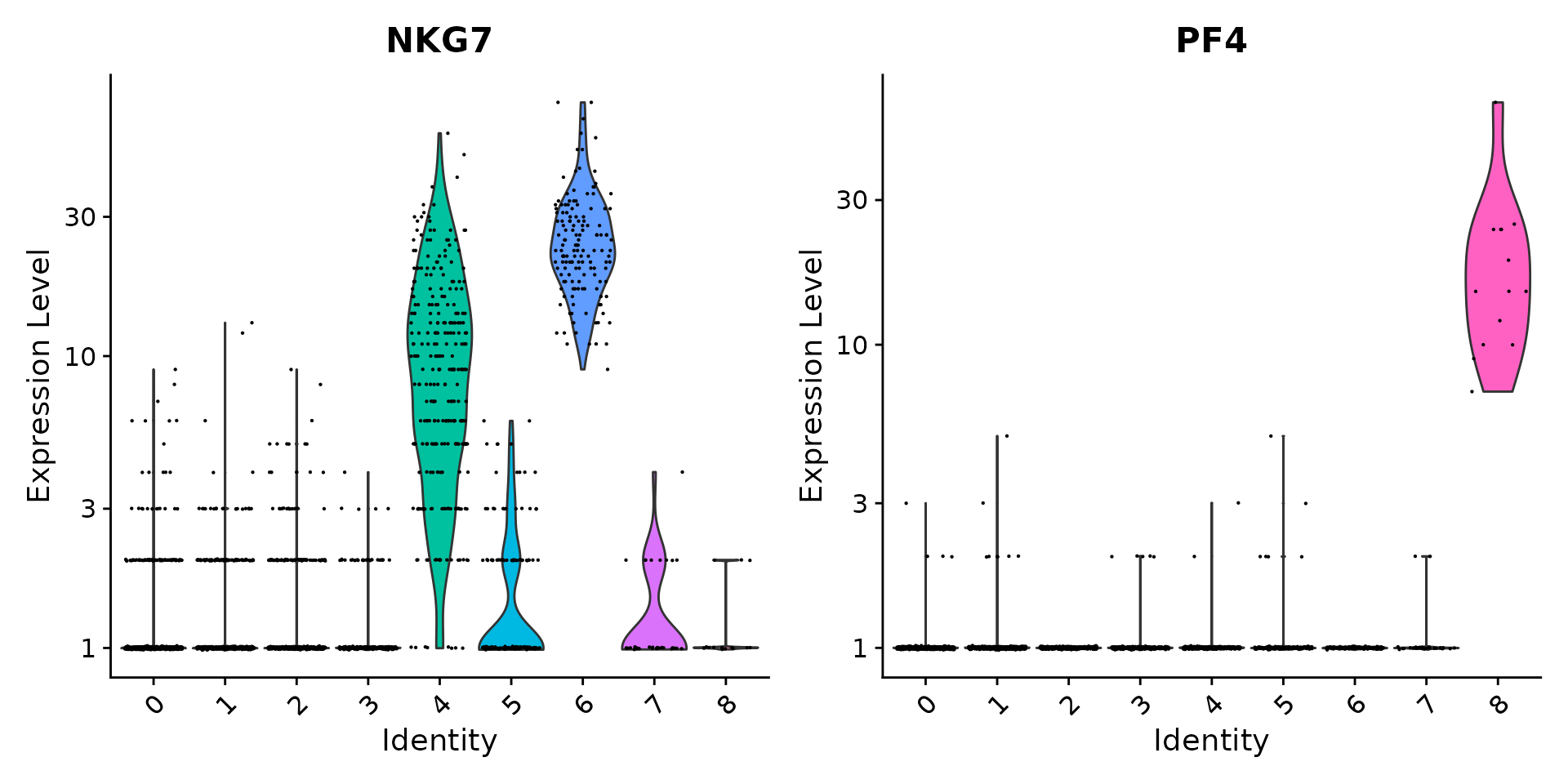
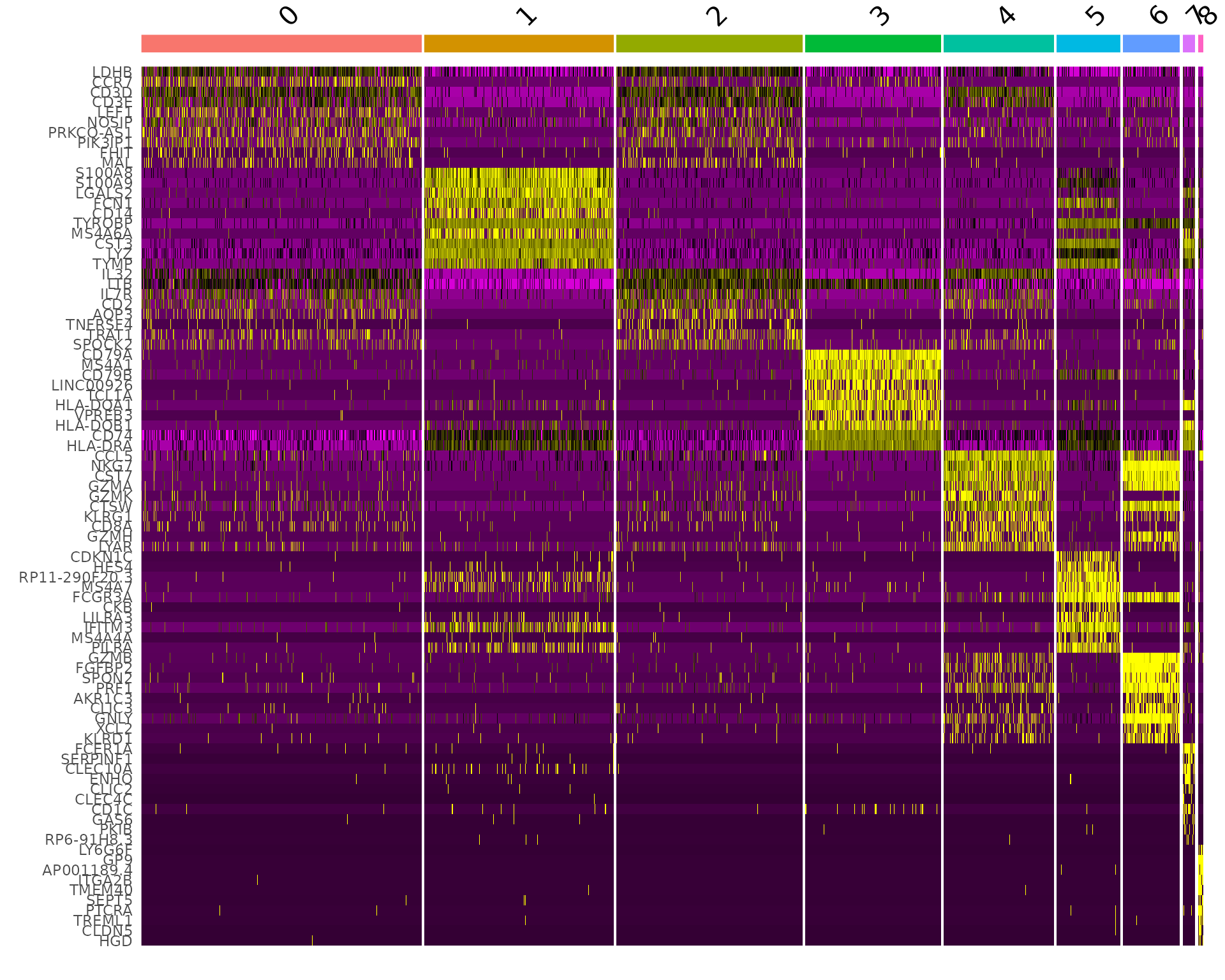
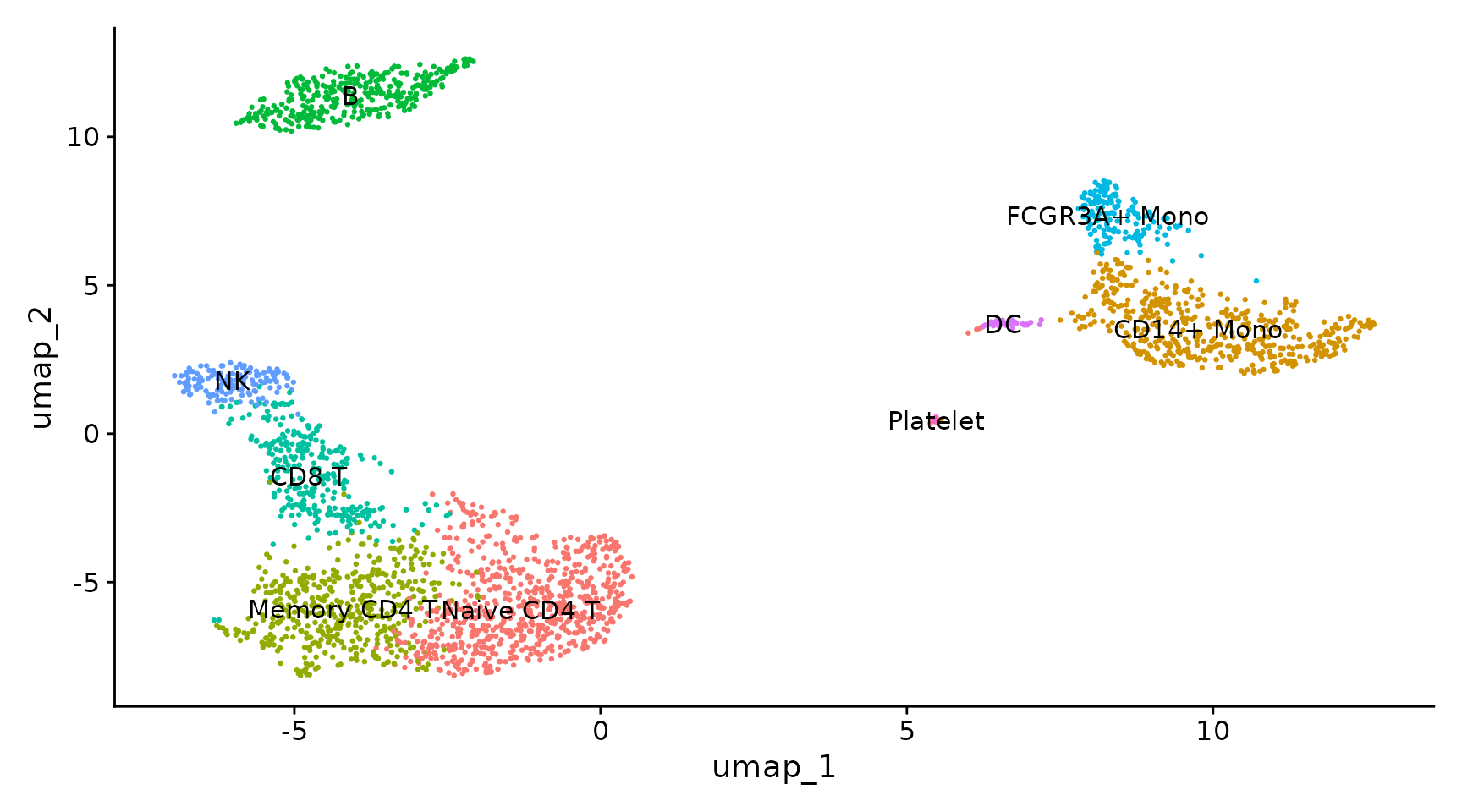
This is my first time to learn siRNA-Seq. The protocol are based on Seurat. Please go and reading more information from Seurat. The codes are derectly copied from Seurat and so, if you are confuzed about my moves, please go to the link below and check by yourselves.
For new users of Seurat, we suggest starting with a guided walk through of a dataset of 2,700 Peripheral Blood Mononuclear Cells (PBMCs) made publicly available by 10X Genomics. This tutorial implements the major components of a standard unsupervised clustering workflow including QC and data filtration, calculation of high-variance genes, dimensional reduction, graph-based clustering, and the identification of cluster markers.
–Seurat
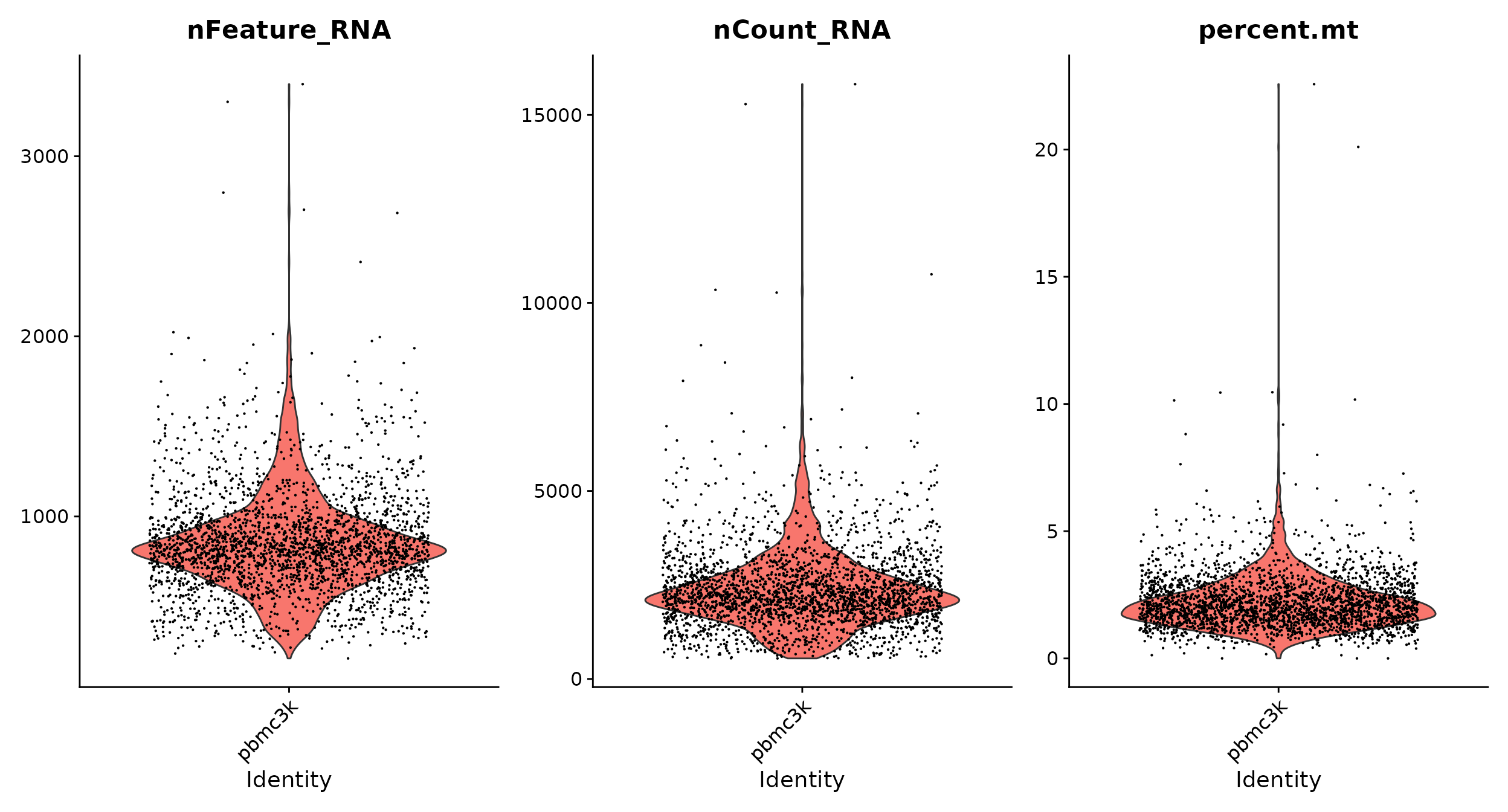
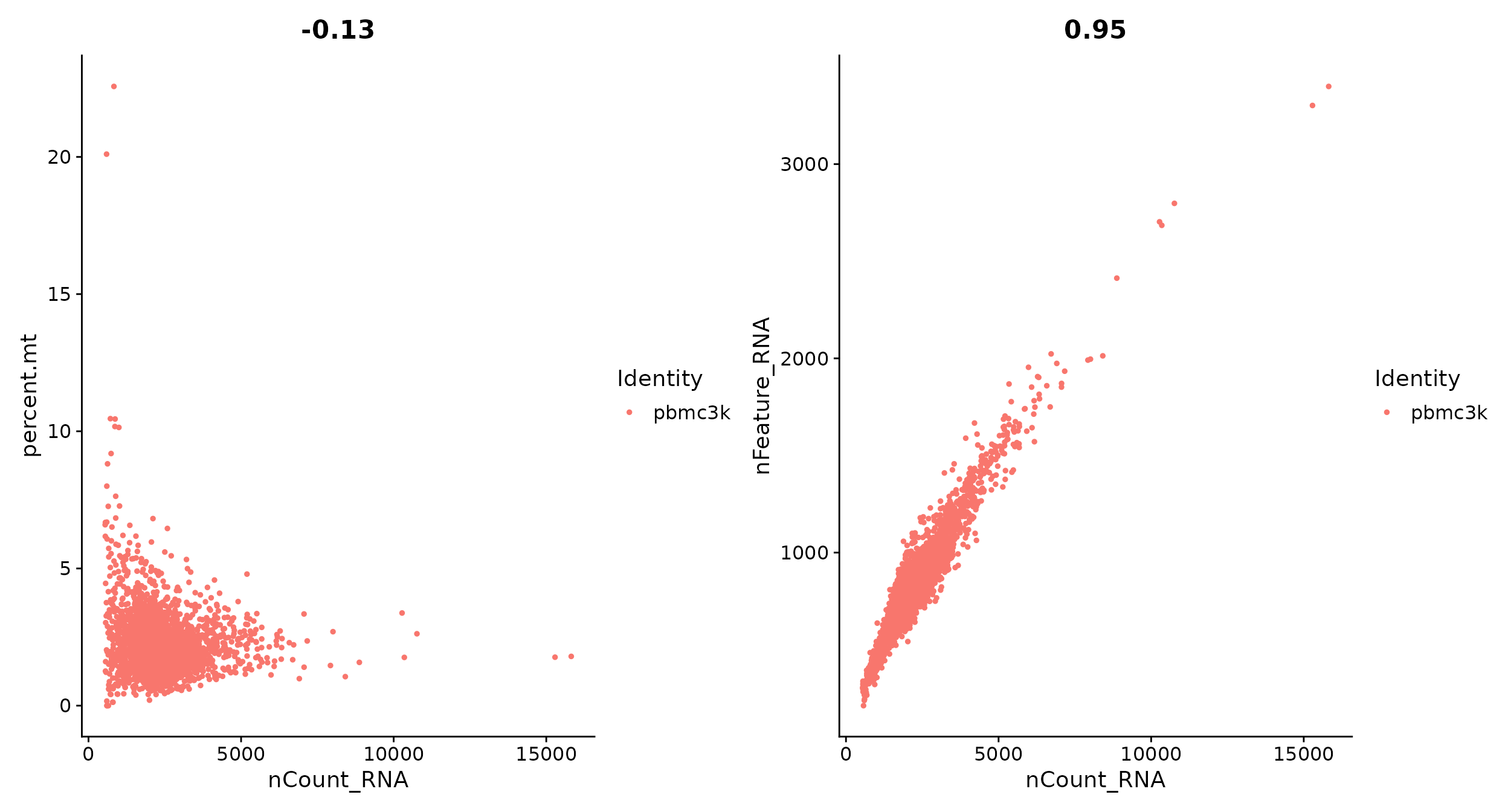
# The [[ operator can add columns to object metadata. This is a great place to stash QC stats pbmc[["percent.mt"]] <- PercentageFeatureSet(pbmc, pattern = "^MT-") VlnPlot(pbmc, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
By default, we employ a global-scaling normalization method “LogNormalize” that normalizes the feature expression measurements for each cell by the total expression, multiplies this by a scale factor (10,000 by default), and log-transforms the result. Normalized values are stored in pbmc[[“RNA”]]@data.
# Identify the 10 most highly variable genes top10 <- head(VariableFeatures(pbmc), 10)
# plot variable features with and without labels plot1 <- VariableFeaturePlot(pbmc) plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE) plot1 + plot2
Scaling the data
Next, we apply a linear transformation (‘scaling’) that is a standard pre-processing step prior to dimensional reduction techniques like PCA. The ScaleData() function:
Shifts the expression of each gene, so that the mean expression across cells is 0
Scales the expression of each gene, so that the variance across cells is 1
This step gives equal weight in downstream analyses, so that highly-expressed genes do not dominate
The results of this are stored in pbmc[["RNA"]]@scale.data
all.genes <- rownames(pbmc) pbmc <- ScaleData(pbmc, features = all.genes)
Why The order of the Normalization → Variable Finding → Scaling matters
Normalization: This step is necessary to account for differences in sequencing depth across cells. Without normalization, cells with more total reads would dominate any downstream analysis.
Finding Variable Features: The primary goal of this step is to identify genes that vary significantly across cells, as these genes can be informative for clustering or other downstream analyses. If you scale the data before this step, you would be artificially inflating the variance of lowly expressed genes and diminishing the variance of highly expressed ones, which might impact the accuracy of variable feature detection.
Scaling: Scaling is performed on the variable features to ensure that they have a mean of 0 and a standard deviation of 1. This step is crucial for dimensionality reduction techniques like PCA, where you don’t want the magnitude of gene expression values to influence the results. If a gene has a high magnitude of expression (let’s say in the thousands) compared to another gene (with values in the tens), without scaling, the gene with higher magnitude values would disproportionately influence the PCA, even if its variance across cells is not particularly informative.
The reason ScaleData is performed after FindVariableFeatures is to ensure that the detection of variable features is based on the actual biological variance in the data, not on the artificial variance introduced by scaling. Once variable features are identified based on their true variance, then we scale them so that no single gene dominates the downstream analyses due to its magnitude.
In simpler terms:
We want to identify variable features based on “real” biological differences, not differences introduced by scaling.
Once we’ve identified the truly variable features, we scale them so that each has an equal opportunity to influence downstream analyses like PCA or clustering, regardless of its absolute expression level.
Scaling before identifying variable features might lead to a situation where genes that aren’t truly variable (in a biological sense) are given undue importance in the analysis.
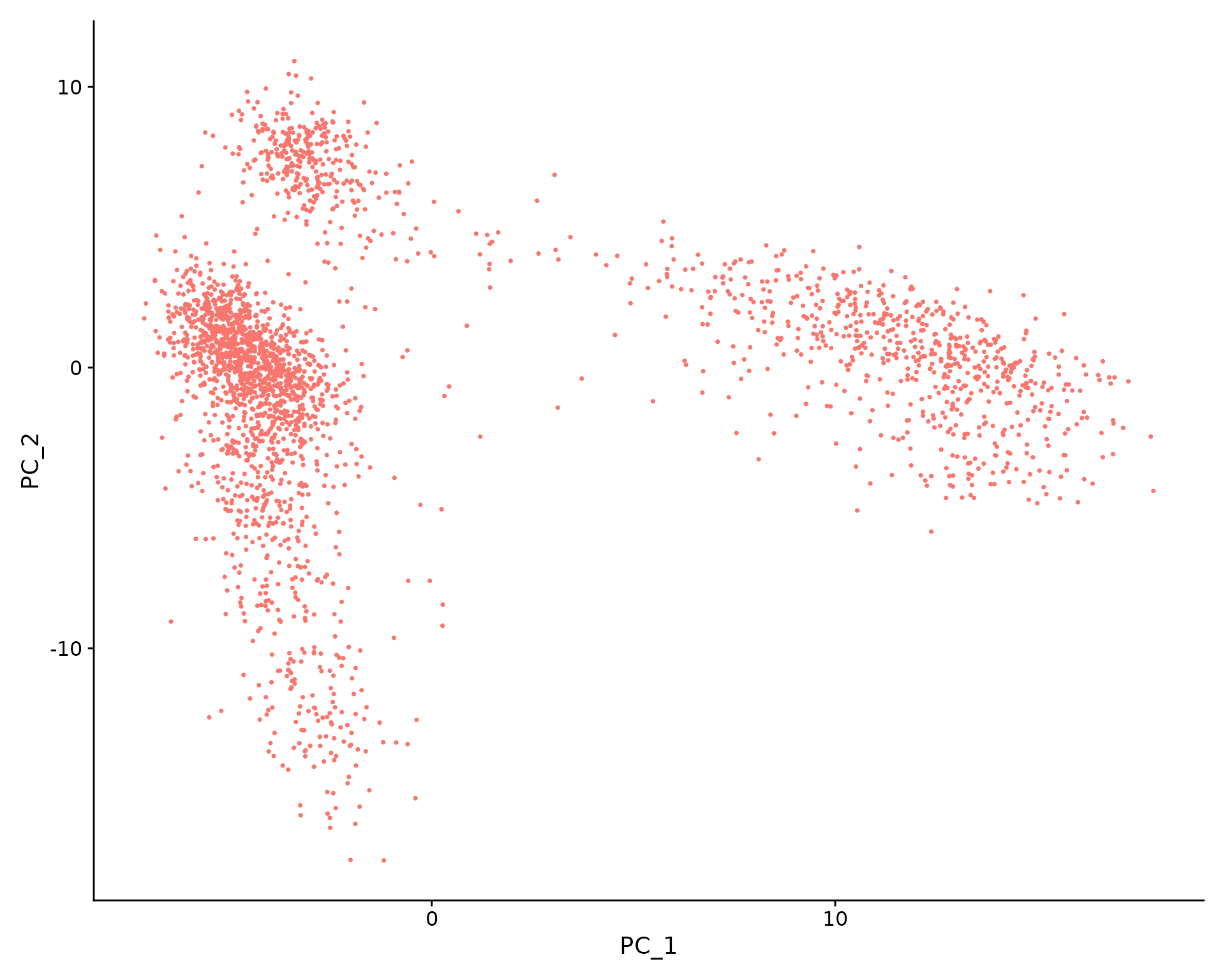
Perform linear dimensional reduction
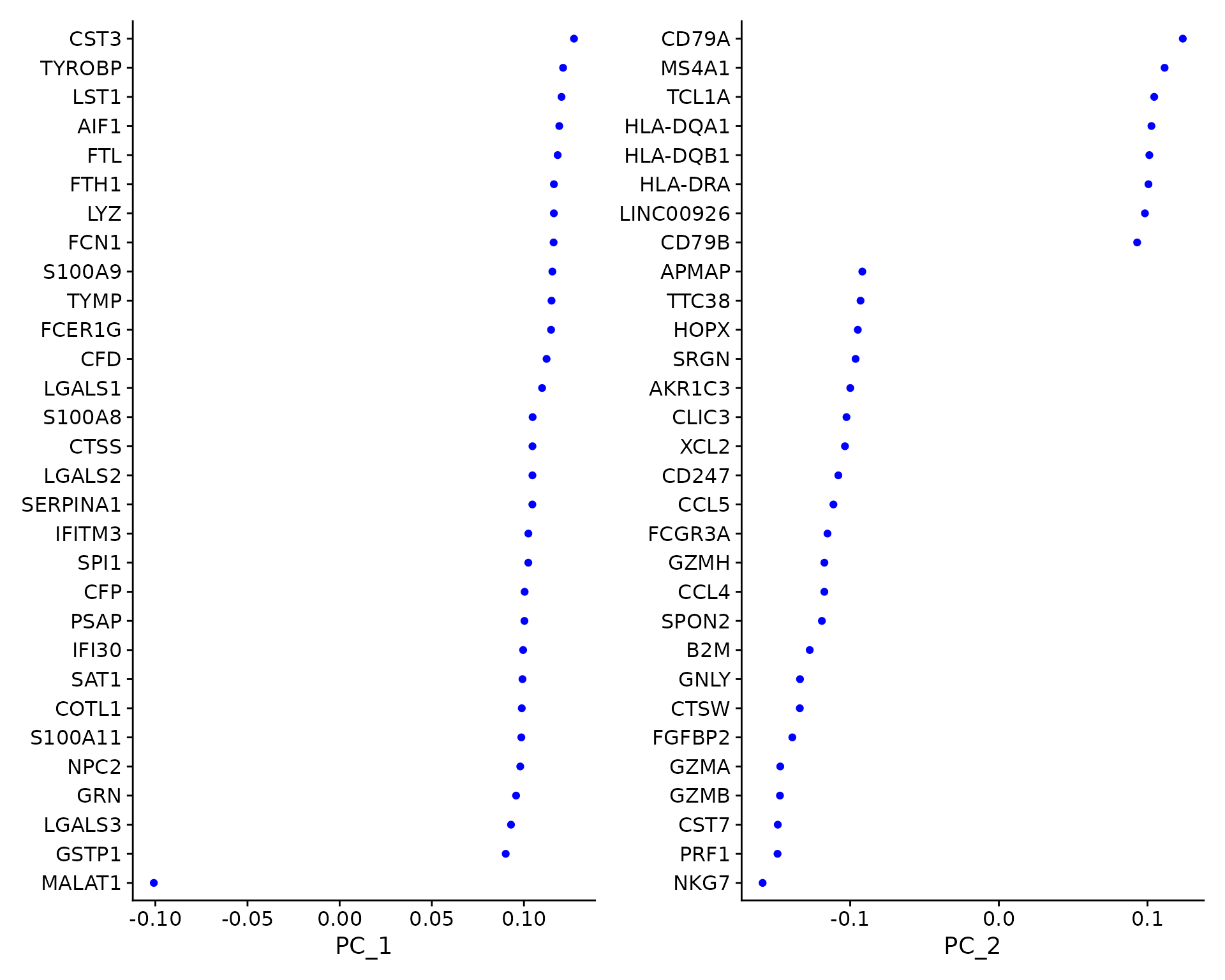
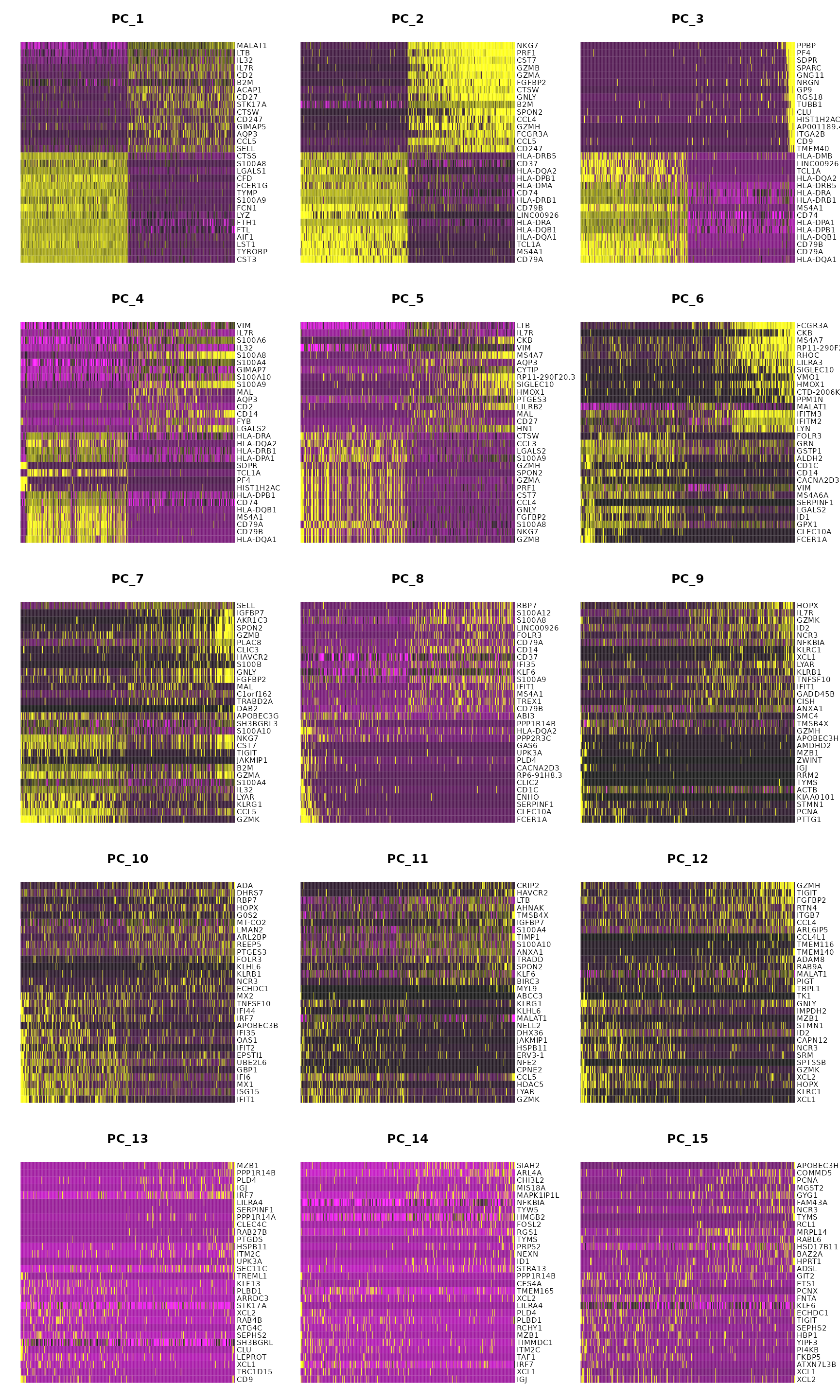
pbmc <- RunPCA(pbmc, features = VariableFeatures(object = pbmc)) # Examine and visualize PCA results a few different ways print(pbmc[["pca"]], dims = 1:5, nfeatures = 5)
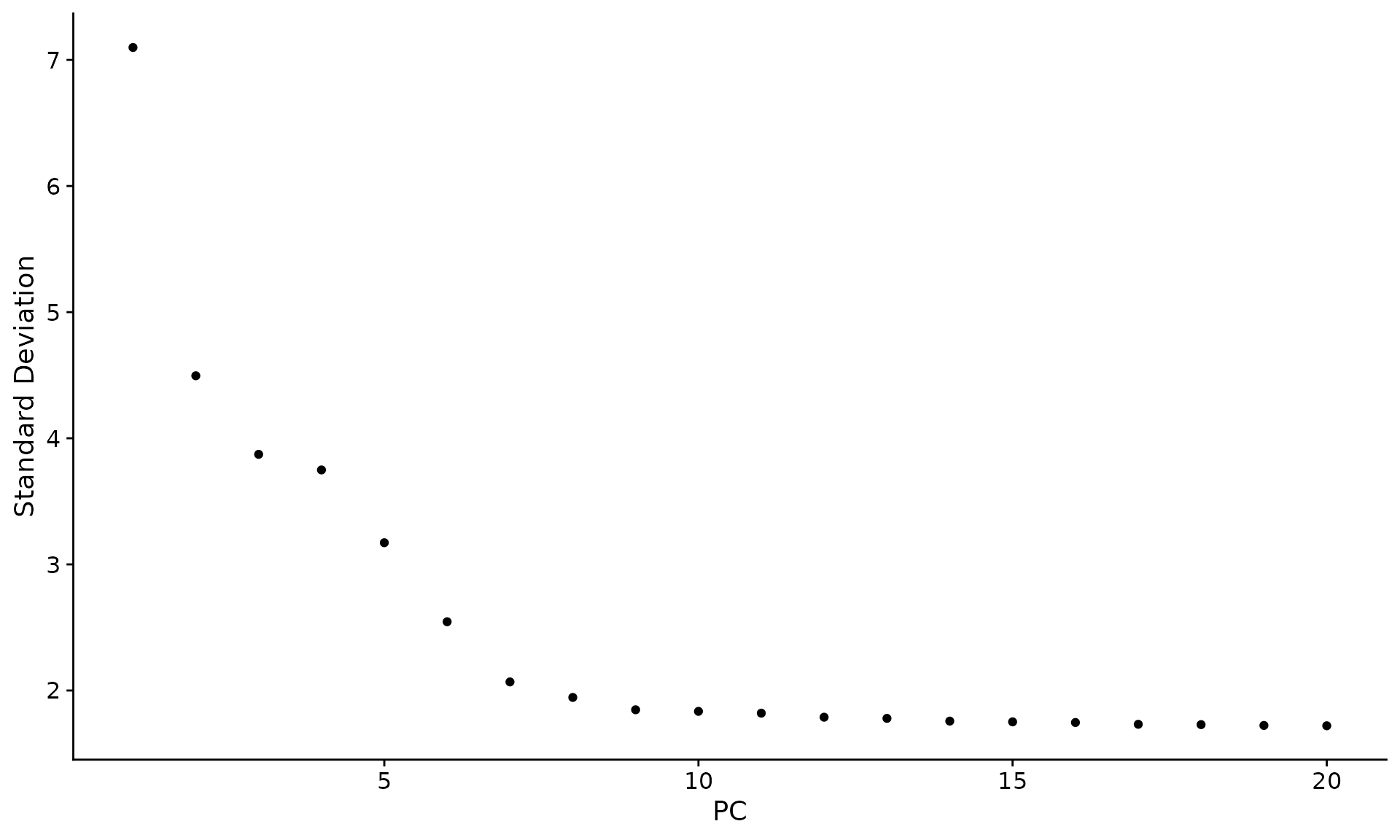
# NOTE: This process can take a long time for big datasets, comment out for expediency. More # approximate techniques such as those implemented in ElbowPlot() can be used to reduce # computation time pbmc <- JackStraw(pbmc, num.replicate = 100) pbmc <- ScoreJackStraw(pbmc, dims = 1:20) JackStrawPlot(pbmc, dims = 1:15)
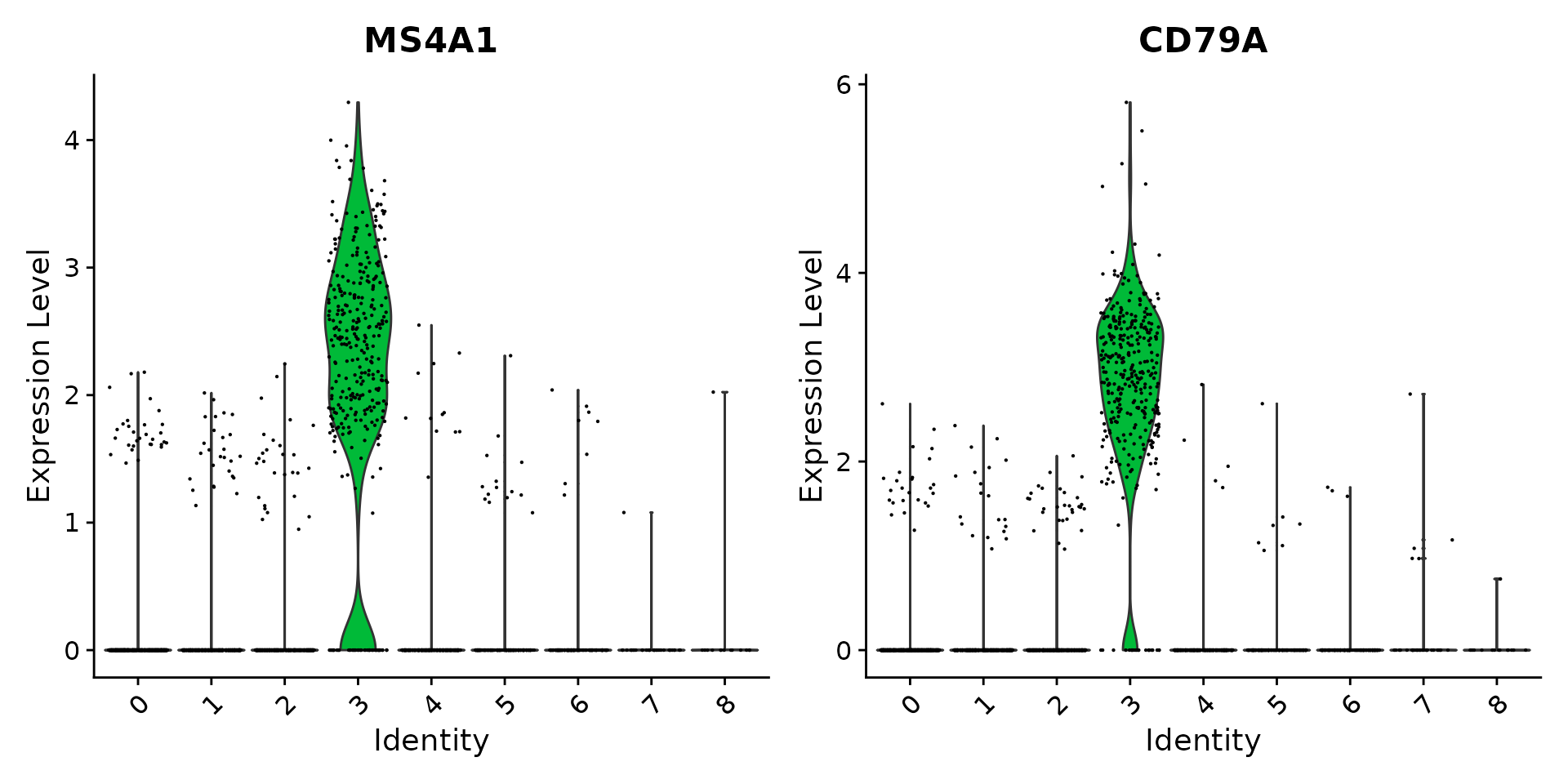
# Look at cluster IDs of the first 5 cells head(Idents(pbmc), 5)
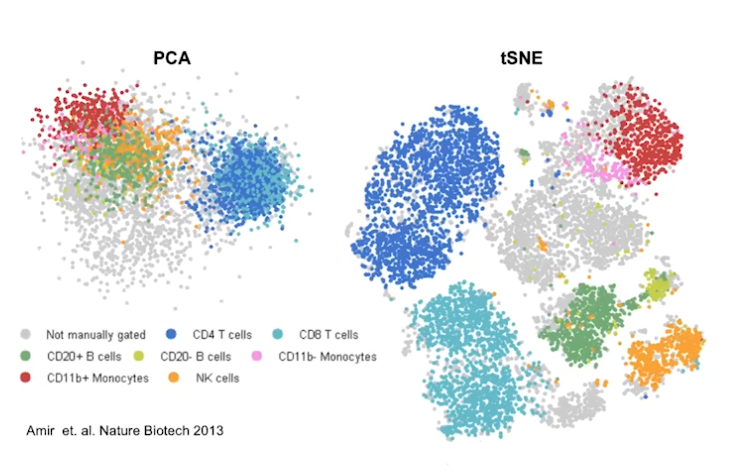
Run non-linear dimensional reduction (UMAP/tSNE)
# If you haven't installed UMAP, you can do so via reticulate::py_install(packages = # 'umap-learn') pbmc <- RunUMAP(pbmc, dims = 1:10) DimPlot(pbmc, reduction = "umap") saveRDS(pbmc, file = "../output/pbmc_tutorial.rds")