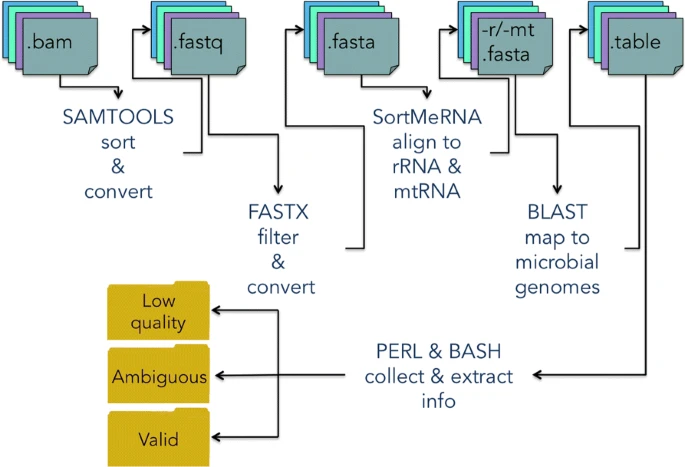
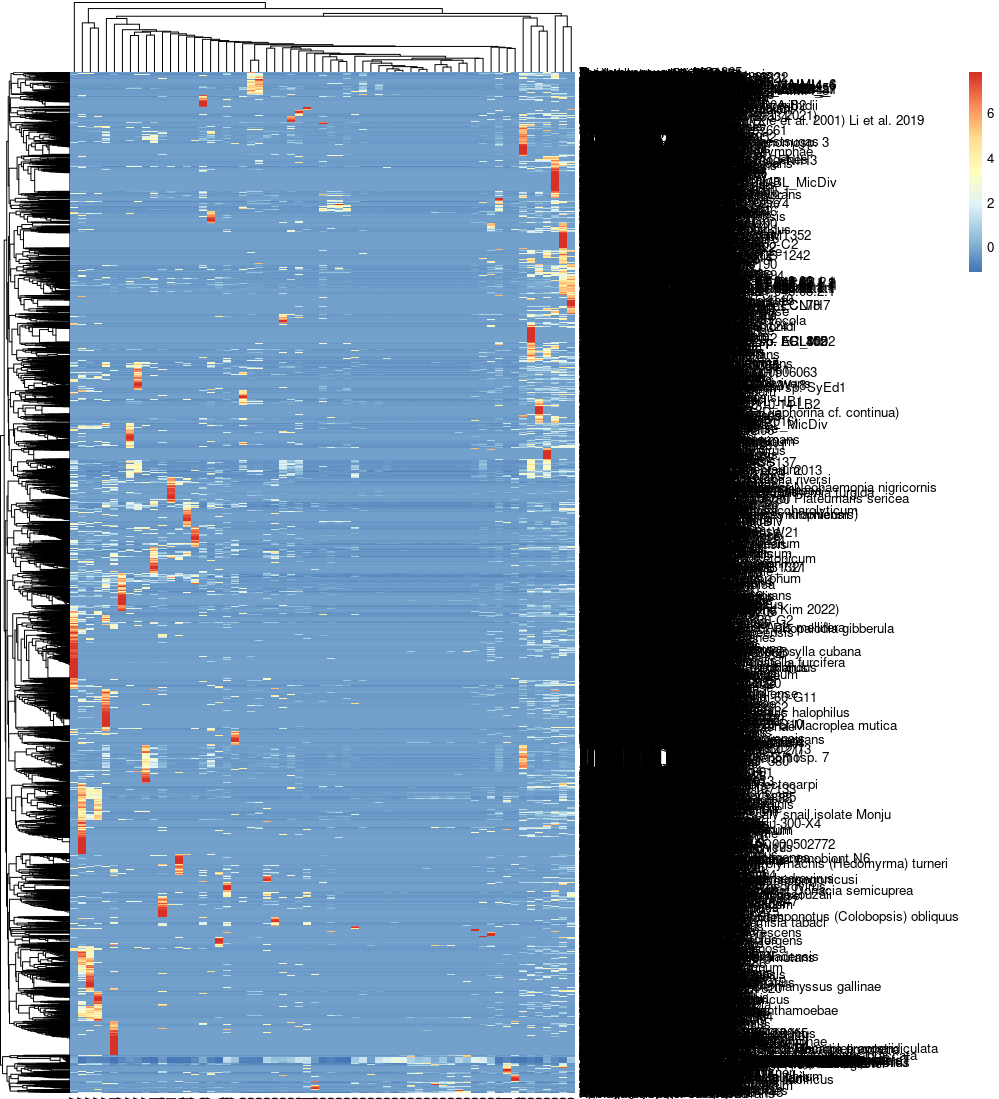
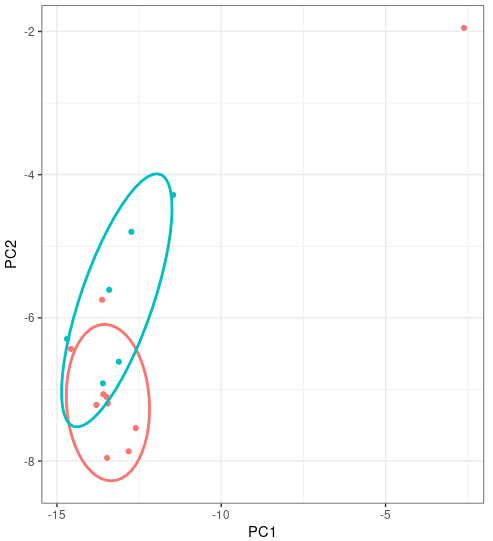
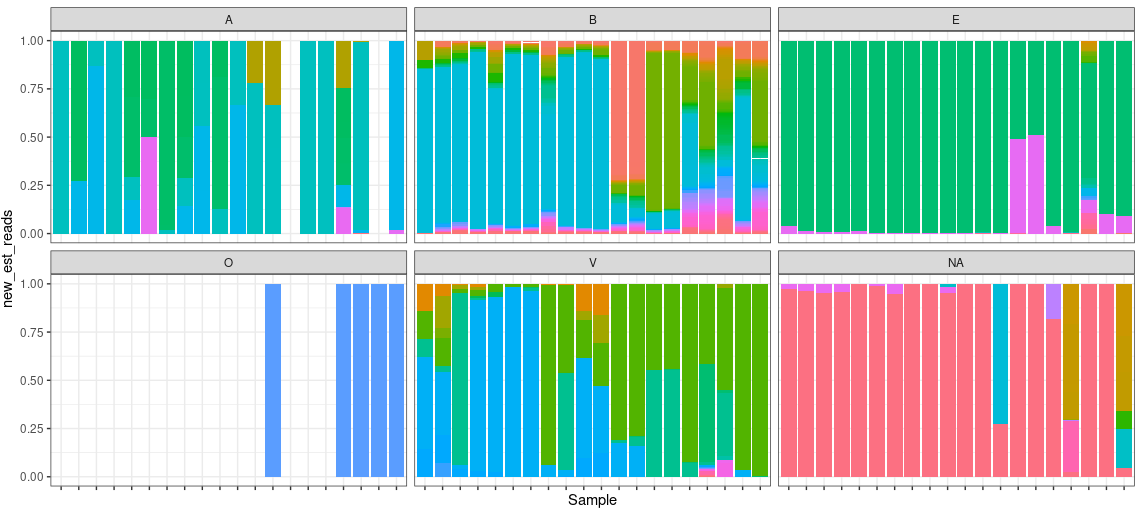
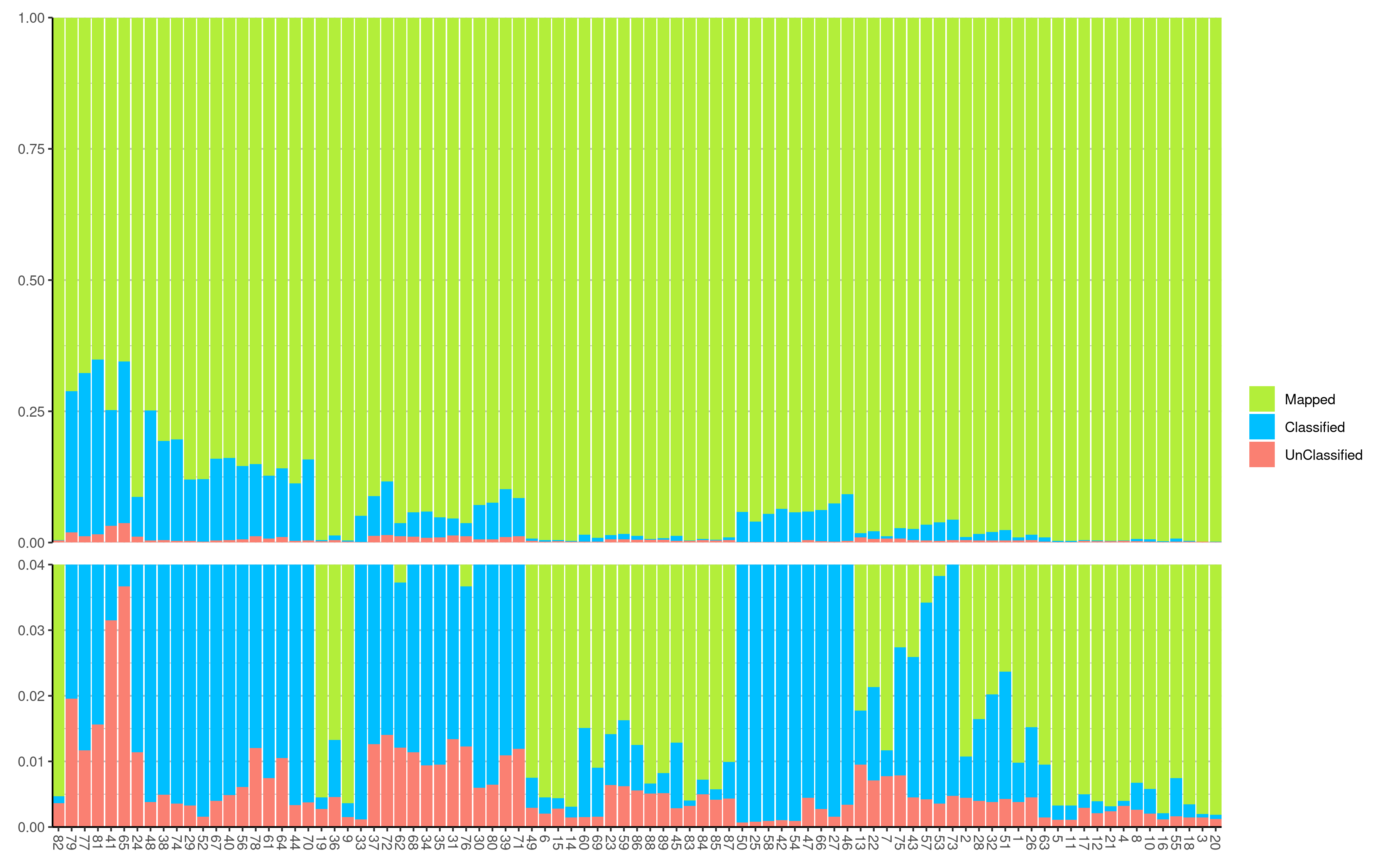
Though we expect that the microbes are free in normal/healthy animal tissue, organ, or brain, we still get a few unmapped reads from NGS. And most of them are contributed by microbes. A Gouin, et al., 2014[1] studied those unmapped reads from 33 plants and believed that those sequences may contribute to the divergence between host plant specialized biotypes.
To identify those unmapped reads, lots of pipelines and tools are developed. Mara Sangiovanni, 2019[2] published the DecontaMiner for investigating the presence of contamination. Zifan Zhu, 2019[3] released MicroPro to study the know and unknown microbes based on the unmapped reads.
Not only in DNA-seq, but RNA-seq could also find lots of unmapped reads and they could be meaningful, too. Artur Gurgul, et al., 2022 [4] found those unmapped reads not only comes from contamination but also come from the hosts and could be used to refine the reference genome. For example, Majid Kazemian, 2015[5] find lots of new or abnormal transcripts from human cancer RNA-seq. A few de novo contigs from unmapped reads could not align to humans but other few kinds of Primates have positive effects on the tumor cell.
Possible sources of unmapped reads:
Biology Issue
Human (From the technician)
symbionts/pathogens
Viruses
Bacteria
fungi
other parasites (exp, mites)
repetitive elements (some alignment tools would ignore them)[4:1]
Once we realized the existence and significance of unmapped reads, we need to find a way to annotate and analyze them. Kraken2 and bracken could be good choose.
Host: 33 pea aphid
Host Genome; mitochondrial genome; primary bacterial symbiont; several secondary symbiont genomes reported for the pea aphid.
Main Steps
Software
Function
Find unmapped reads
Bowtie2
align to Reference Genome
Find unmapped reads
Samtools
extract unmaped reads
Unmapped reads assembly
Prinseq
trimme the quality below 20 within a window of 10 nucleotides, and only sequences of at least 66 nucleotides in length were retained
Unmapped reads Analysis
Compareads
Find similar reads between two sets of reads in an assembly-free manner
Unmapped reads assembly
ABySS
Unmapped reads de novo assembling
Unmapped reads assembly
Bowtie2
align unmapped reads to assembled Contigs
Contigs analysis
BLASTClust
Found homologous large contigous.
Contigs analysis
BLASTn
Found homologous contigous
Unmapped reads assembly
readFilter
only reads for which 68% of the length was covered by 31-mers present at least 100 times in the data set were retained
Unmapped reads assembly
SPAdes
De novo assembly
Potentially divergent
Mummer
Global aligner, against reference: >80% identity and >500 bp were retained
Contigs analysis
GeneMarkS+
Predict the protein from de novo contigs.
Contigs analysis
Blastp
Predicted protein annotation
Potentially divergent
Stampy
An aligner performs well when divergent is high.
Potentially divergent
GATK
SNP calling
PS:
Why Bowtie2: the percentage of unmapped reads was higher than for Bowtie2 (on average over the 33 individuals, 6.1% vs 3.7% for BWA and Bowtie2, respectively)
Jen Lu; 2017: If a 150bp read is 100% identical to two different species, Kraken will assign it to their lowest common ancestor (LCA), which could be at the genus level or higher. But some reads within a particular sample usually come from the unique (or species-specific) portion of the genome. Bracken uses this information, plus information about the similarity between the sister species, to “push down” reads from the genus level to the species level.
Lu J., et al. showed that comparing to QiiMe, Kraken2 has uncompetable speed in annotation and the results from kraken-bracken are even better than QiiMe[8]. I think it’s time for QiiMe move a room for Kraken2 now.
Technically, you can find the unmapped reads by samtools view *.bam| awk '$3=="*"'. But I don’t know why the number of reads is less than the result from samtools stats. So, the easiest way to achieve this is:
In this code, reads would be split into 3 files: 1.fq.gz, 2.fq.gz, and s.fq.gz. The first two are paired reads and the 3rd is unpaired reads. In Kraken classification under paired parameter, unpaired reads in fq file would be ignored. So, we need to store them independently and run kraken twice.
If some samples are single ends, the result cannot be stored with this command. But the reads would be printed into log file: Log/*.out. So, we can compress it into fq.gz
# Paired for SAMPLE in $(ls $FQ_DB/*_1.fq.gz| sed "s=$FQ_DB/==;s/_[12].fq.gz//"); do if [ -f $SAMPLE.P.sorted.bam ]; then echo$SAMPLE is done else echo$SAMPLE can not be find sed "s/Hi/$SAMPLE\_P_$DB/" Model.sh > script/$SAMPLE\_P_$DB.sh echo bwa mem $DB_FA$FQ_DB/$SAMPLE\_1.fq.gz $FQ_DB/$SAMPLE\_1.fq.gz -t 64 \|samtools view -S -b -\|samtools sort \> $SAMPLE.P.sorted.bam >> script/$SAMPLE\_P_$DB.sh echo samtools index $SAMPLE.P.sorted.bam >> script/$SAMPLE\_P_$DB.sh sbatch script/$SAMPLE\_P_$DB.sh fi done
# Single for SAMPLE in $(ls $FQ_DB/*_s.fq.gz| sed "s=$FQ_DB/==;s/_s.fq.gz//"); do if [ -f $SAMPLE.S.sorted.bam ]; then echo$SAMPLE is done else echo$SAMPLE can not be find Clean echo bwa mem $DB_FA$FQ_DB/$SAMPLE\_s.fq.gz -t 64 \|samtools view -S -b -\|samtools sort \> $SAMPLE.S.sorted.bam >> script/$SAMPLE\_S_$DB.sh echo samtools index $SAMPLE.S.sorted.bam >> script/$SAMPLE\_S_$DB.sh sbatch script/$SAMPLE\_S_$DB.sh fi done
## Check unexpected size of file rm $(du *.sorted.bam | awk '$1<=100'| awk '{print $2}') ## After removed failed files, we can excute the for loop again #. #. #. ## finally, we can mv all bam into another directory mv *.sorted.* $OUTPUT
Counting results
There are two bam files. One is paired result, another is single end result. For avoid counting them twice in paired result, we can samply sort and uniq them. The only problem is it would consume lots of memory and time.
for SAMPLE in $(ls Bam_clean/*.bam| sed 's/.[PS].sorted.bam//;s=Bam_clean/=='| sort |uniq); do sed "s/Hi/$SAMPLE_Clean_count/" Model.sh > script/$SAMPLE.bam_count.sh echo"cat Bam_clean/$SAMPLE.[PS].sorted.bam| samtools view| awk '{print $1,$3}'| awk -F"_" '{print $1}'| sort|uniq| awk '{print $2}'|sort| uniq -c |sed 's/^ *//'> Bam_Stats/$SAMPLE.Clean.csv" >> script/$SAMPLE.bam_count.sh sbatch script/$SAMPLE.bam_count.sh done
extract unmapped reads again
Codes folded for extract reads again
module load samtools
BAM=Bam_clean OUT_dir=FQ_split
mkdir $OUT_dir script Log
## paired-end reads for i in $(ls $BAM/*.P.sorted.bam); do SAMPLE=$(echo$i| awk -F"/"'{print $NF}'| sed "s/.P.sorted.bam//"); if [ -f $OUT_dir/$SAMPLE\_1.fq.gz ]; thenecho$SAMPLE is done else sed "s/Hi/$SAMPLE/" Model.sh > script/$SAMPLE\_fq.sh echo"samtools fastq -@ 8 -f 4 $i -1 $OUT_dir/$SAMPLE""_1.fq.gz -2 $OUT_dir/$SAMPLE""_2.fq.gz -s $OUT_dir/$SAMPLE""_s.fq.gz" >> script/$SAMPLE\_fq.sh sbatch script/$SAMPLE\_fq.sh fi done
# Check for Single Ends
for i in $(ls $BAM/*.S.sorted.bam); do SAMPLE=$(echo$i| awk -F"/"'{print $NF}'| sed "s/.S.sorted.bam//"); sed "s/Hi/$SAMPLE/" Model.sh > script/$SAMPLE\_fq.sh echo"samtools fastq -@ 8 -f 4 $i| gzip -f > $OUT_dir/$SAMPLE""_s.fq.gz" >> script/$SAMPLE\_fq.sh sbatch script/$SAMPLE\_fq.sh done
# record unclassified reads for i in $(ls Kraken_result/*.kraken); do OUT=$(echo$i| sed 's=Kraken_result/==;s/.kraken/.uc.list/') grep ^U $i| awk '{print $2}' > Log/$OUT done
Bams_stats is generated by samtools stats SAMPLE.bam > SAMPLE.stats.csv
In Kraken, when you use paired reads, it would only show the number of the paired reads which means you only get a half size of the reads compared to the result from samtools stats. As a result, we need to multiply them by 2 during visualization.
ggsave("img/Number_Bar_classR.png", h= 7.5, w = 12)
Header One
Header Two
Full Taxonomic information based on ID
Some times, we’d like to analyze the Eucariaotic or Viral independently. Them, we need to find the phlums of them based on taxonomic ID. etetoolkit Can help find the phylums efficiently.
Gouin, A., Legeai, F., Nouhaud, P. et al. Whole-genome re-sequencing of non-model organisms: lessons from unmapped reads. Heredity 114, 494–501 (2015). https://doi.org/10.1038/hdy.2014.85↩︎↩︎
Sangiovanni, M., Granata, I., Thind, A. et al. From trash to treasure: detecting unexpected contamination in unmapped NGS data. BMC Bioinformatics 20 (Suppl 4), 168 (2019). https://doi.org/10.1186/s12859-019-2684-x↩︎↩︎
Zhu, Z., Ren, J., Michail, S. et al. MicroPro: using metagenomic unmapped reads to provide insights into human microbiota and disease associations. Genome Biol 20, 154 (2019). https://doi.org/10.1186/s13059-019-1773-5↩︎↩︎
Gurgul, A., Szmatoła, T., Ocłe sources of unmapped reads are various: they may derive from characterized or unchoń, E. et al. Another lesson from unmapped reads: in-depth analysis of RNA-Seq reads from various horse tissues. J Appl Genetics 63, 571–581 (2022). https://doi.org/10.1007/s13353-022-00705-z↩︎↩︎↩︎↩︎↩︎
Kazemian, Majid, et al. “Comprehensive assembly of novel transcripts from unmapped human RNA‐Seq data and their association with cancer.” Molecular systems biology 11.8 (2015): 826. https://doi.org/10.15252/msb.156172↩︎
Laine, V.N., Gossmann, T.I., van Oers, K. et al. Exploring the unmapped DNA and RNA reads in a songbird genome. BMC Genomics 20, 19 (2019). https://doi.org/10.1186/s12864-018-5378-2↩︎
De Bourcy, Charles FA, et al. “A quantitative comparison of single-cell whole genome amplification methods.” PloS one 9.8 (2014): e105585. ↩︎
Lu J, Salzberg S L. Ultrafast and accurate 16S rRNA microbial community analysis using Kraken 2[J]. Microbiome, 2020, 8(1): 1-11. ↩︎