
Chip-Seq, from Know to Unknown = =
CHIP-Seq
General infor: CD_Genome.pdf
| aaaaaaaaaaaaaaaaaaaaaaaaaaaa | |
|---|---|
 © wiki © wiki |
ChIP-sequencing, also known as ChIP-seq, is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation (ChIP) with massively parallel DNA sequencing to identify the binding sites of DNA-associated proteins. It can be used to map global binding sites precisely for any protein of interest. Previously, ChIP-on-chip was the most common technique utilized to study these protein–DNA relations.wiki |
Library preparation
At the day of 2022-09-12, Illumina high light the product: “TruSeq ChIP Library Preparation Kit”. According to their statements, it have a high sensitivity and high compatibility. It requires 5 ng ChIP-derived DNA. Illumina; 2022/09
Data Processing
Illumina; 2022/09 suggests using MACS2 to identify enriched regions, and HOMER to discover motifs within these regions.
Input:
- Treatment Samples
- Control samples
Output: - Interactive Annotated Peak/Motif Explorer
- Alignment files (BAMs)
- Annotation, Peak, and Motif files.
| Pipeline from CD Genomics |
|---|
 |
| Tool | Notes | Web address |
| Short-read aligners | ||
| BWA (Burrows-Wheeler Aligner) | Fast and efficient; based on the Burrows-Wheeler transform | http://bio-bwa.sourceforge.net |
| Bowtie | Similar to BWA, part of suite of tools that includes TopHat and CuffLinks for RNA-seq processing | http://bowtie-bio.sourceforge.net |
| GSNAP (Genomic Short-read Nucleotide Alignment Program) | Considers a set of variant allele inputs to better align to heterozygous sites | http://research-pub.gene.com/gmap |
| Wikipedia list of aligners | A comprehensive list of available short-read aligners, with descriptions and links to download the software | http://en.wikipedia.org/wiki/List_of_ sequence_alignment_software#Short- Read_Sequence_Alignment |
| Peak callers | ||
| MACS (Model-based Analysis for ChIP-seq) | Fits data to a dynamic Poisson distribution; works with and without control data | http://liulab.dfci.harvard.edu/MACS |
| PeakSeq | Takes into account differences in mappability of genomic regions; enrichment based on FDR (false-discovery rate) calculation | http://info.gersteinlab.org/PeakSeq |
| ZINBA (Zero-Inflated Negative Binomial Algorithm) | Can incorporate multiple genomic factors, such as mappability and GC content; can work with point-source and broad-source peak data | http://code.google.com/p/zinba |
Pipeline from EncodeProject
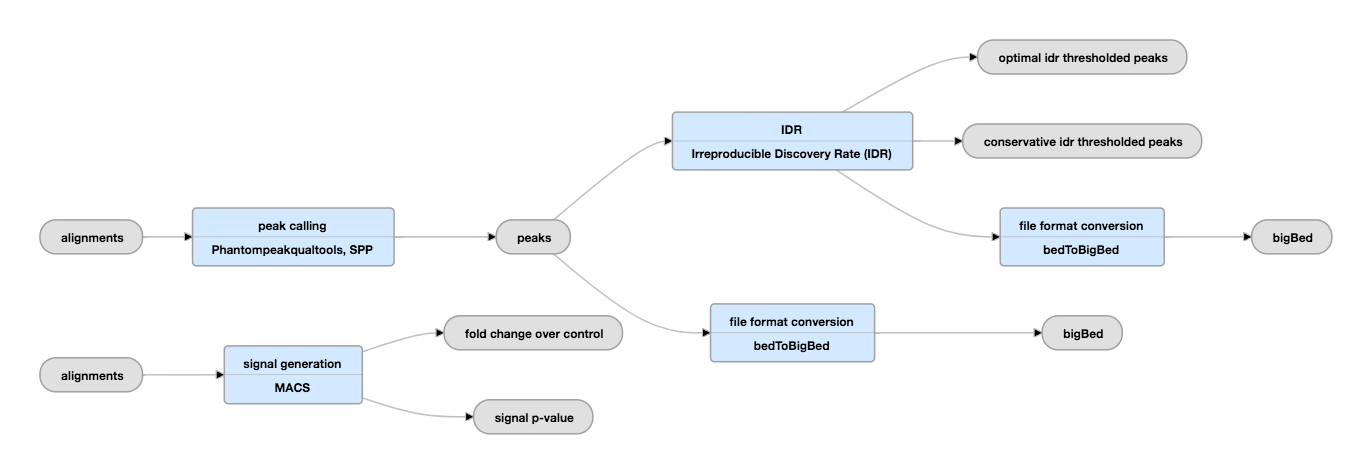
To be notice
EncodeProject list a different pipeline for treating Hitone ChiP-seq and Transcription Factor ChiP-seq.
Why different:
If you’re looking at transcription factors, then you should use a peak finder like MACS (http://liulab.dfci.harvard.edu/MACS/) to search for enrichment of your TF compared to a background samples such as input or no-ab control.
If however, you are profiling a fairly well characterised histone mark such as H3K9ac or H3K4me3 then you can simply count the reads that map near the promoters as a proxy for gene activation/repression. This can be done with Bedtools,
© Mark Ziemann, 2015
Tutorials
General Information: crazyhottommy; 2015
Tutorial from simonvh, 2018
Tutorial from IGR-lab, 2022
Tutorial from 生信技能树, 2018
Detailed Tutorial from jmzeng1314, 2018
Post analysis by R package: EpigeneticsCSAMA; 2016
More post analysis with bioconductor: brc from rockefeller edu, 2021
Example of ChiP-seq reports from commercial
Part 2; Practice
A quick Tutorial from 宸宇卿
Source:
Environment Settel
- sra-toolkit: prepare data from NCBI
|
Data Preparation
- Prepare directories and Dowload test data
|
fastpto remove the header and and low quality reads andfastqcto check the quality of the reads, agian.
|
In for loop:
|
- Reference Genome
|
Map the reads into reference genome
By following steps from above, you should get directories like below. Quality trimmed reads are in Fastp. Since they are single ends, we can easily align them into reference genome with less codes then paired one.
. ├── Fastp │ ├── SRR1042593_1.fastq │ ├── SRR1042594_1.fastq │ ├── SRR1042595_1.fastq │ ├── SRR1042596_1.fastq │ ├── SRR1042597_1.fastq │ ├── SRR1042598_1.fastq │ ├── SRR1042599_1.fastq │ └── SRR1042600_1.fastq ├── FastQC ├── hg19 └── Raw_fq
|
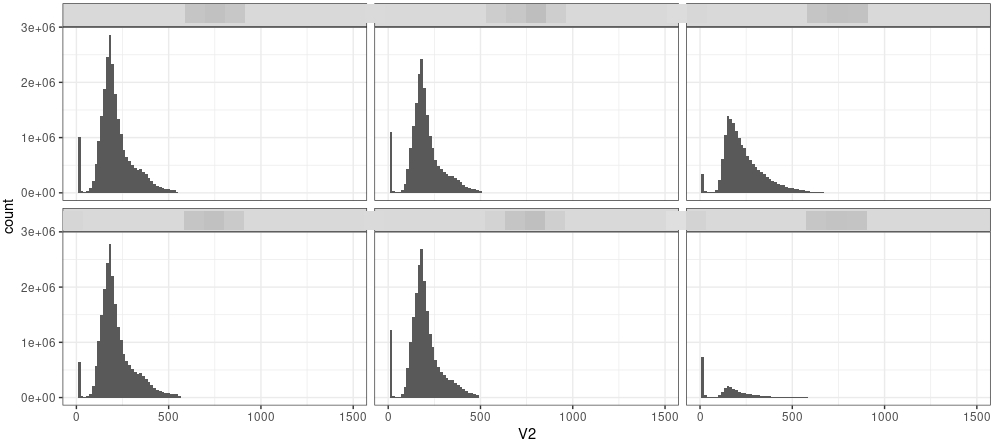
Insert size.
 |
|---|
An example of insert size for Chip-seq result.
MACS: peak calling
Documentation: macs3-project/MACS; github
|
-g
size of the genome. It can be 1.0e+9 or 1000000000, or shortcuts:'hs' for human (2.7e9), 'mm' for mouse (1.87e9), 'ce' for C. elegans (9e7) and 'dm' for fruitfly (1.2e8), Default:hs
Results:
.
├── test_control_lambda.bdg
├── test_model.r
├── test_peaks.narrowPeak
├── test_peaks.xls
├── test_summits.bed
└── test_treat_pileup.bdg
Output Log:
INFO @ Mon, 26 Sep 2022 11:33:39: # Command line: callpeak -t Bam/SRR1042593.sorted.bam -c Bam/SRR1042598.sorted.bam -f BAM -g hs -n test -B -q 0.01 # ARGUMENTS LIST: # name = test # format = BAM # ChIP-seq file = ['Bam/SRR1042593.sorted.bam'] # control file = ['Bam/SRR1042598.sorted.bam'] # effective genome size = 2.70e+09 # band width = 300 # model fold = [5, 50] # qvalue cutoff = 1.00e-02 # The maximum gap between significant sites is assigned as the read length/tag size. # The minimum length of peaks is assigned as the predicted fragment length "d". # Larger dataset will be scaled towards smaller dataset. # Range for calculating regional lambda is: 1000 bps and 10000 bps # Broad region calling is off # Paired-End mode is off INFO @ Mon, 26 Sep 2022 11:33:39: #1 read tag files... INFO @ Mon, 26 Sep 2022 11:33:39: #1 read treatment tags... INFO @ Mon, 26 Sep 2022 11:33:41: 1000000 reads parsed . . . INFO @ Mon, 26 Sep 2022 11:35:12: 50000000 reads parsed INFO @ Mon, 26 Sep 2022 11:35:19: 50255304 reads have been read. INFO @ Mon, 26 Sep 2022 11:35:19: #1 tag size is determined as 51 bps INFO @ Mon, 26 Sep 2022 11:35:19: #1 tag size = 51.0 INFO @ Mon, 26 Sep 2022 11:35:19: #1 total tags in treatment: 16355294 INFO @ Mon, 26 Sep 2022 11:35:19: #1 user defined the maximum tags... INFO @ Mon, 26 Sep 2022 11:35:19: #1 filter out redundant tags at the same location and the same strand by allowing at most 1 tag(s) INFO @ Mon, 26 Sep 2022 11:35:20: #1 tags after filtering in treatment: 9821254 INFO @ Mon, 26 Sep 2022 11:35:20: #1 Redundant rate of treatment: 0.40 INFO @ Mon, 26 Sep 2022 11:35:20: #1 total tags in control: 50255304 INFO @ Mon, 26 Sep 2022 11:35:20: #1 user defined the maximum tags... INFO @ Mon, 26 Sep 2022 11:35:20: #1 filter out redundant tags at the same location and the same strand by allowing at most 1 tag(s) INFO @ Mon, 26 Sep 2022 11:35:20: #1 tags after filtering in control: 47926382 INFO @ Mon, 26 Sep 2022 11:35:20: #1 Redundant rate of control: 0.05 INFO @ Mon, 26 Sep 2022 11:35:20: #1 finished! INFO @ Mon, 26 Sep 2022 11:35:20: #2 Build Peak Model... INFO @ Mon, 26 Sep 2022 11:35:20: #2 looking for paired plus/minus strand peaks... INFO @ Mon, 26 Sep 2022 11:35:23: #2 Total number of paired peaks: 5877 INFO @ Mon, 26 Sep 2022 11:35:23: #2 Model building with cross-correlation: Done INFO @ Mon, 26 Sep 2022 11:35:23: #2 finished! INFO @ Mon, 26 Sep 2022 11:35:23: #2 predicted fragment length is 211 bps INFO @ Mon, 26 Sep 2022 11:35:23: #2 alternative fragment length(s) may be 211 bps INFO @ Mon, 26 Sep 2022 11:35:23: #2.2 Generate R script for model : test_model.r INFO @ Mon, 26 Sep 2022 11:35:23: #3 Call peaks... INFO @ Mon, 26 Sep 2022 11:35:23: #3 Pre-compute pvalue-qvalue table... INFO @ Mon, 26 Sep 2022 11:37:56: #3 In the peak calling step, the following will be performed simultaneously: INFO @ Mon, 26 Sep 2022 11:37:56: #3 Write bedGraph files for treatment pileup (after scaling if necessary)... test_treat_pileup.bdg INFO @ Mon, 26 Sep 2022 11:37:56: #3 Write bedGraph files for control lambda (after scaling if necessary)... test_control_lambda.bdg INFO @ Mon, 26 Sep 2022 11:37:56: #3 Pileup will be based on sequencing depth in treatment. INFO @ Mon, 26 Sep 2022 11:37:56: #3 Call peaks for each chromosome... INFO @ Mon, 26 Sep 2022 11:39:10: #4 Write output xls file... test_peaks.xls INFO @ Mon, 26 Sep 2022 11:39:10: #4 Write peak in narrowPeak format file... test_peaks.narrowPeak INFO @ Mon, 26 Sep 2022 11:39:10: #4 Write summits bed file... test_summits.bed INFO @ Mon, 26 Sep 2022 11:39:10: Done!
Visual
IGV
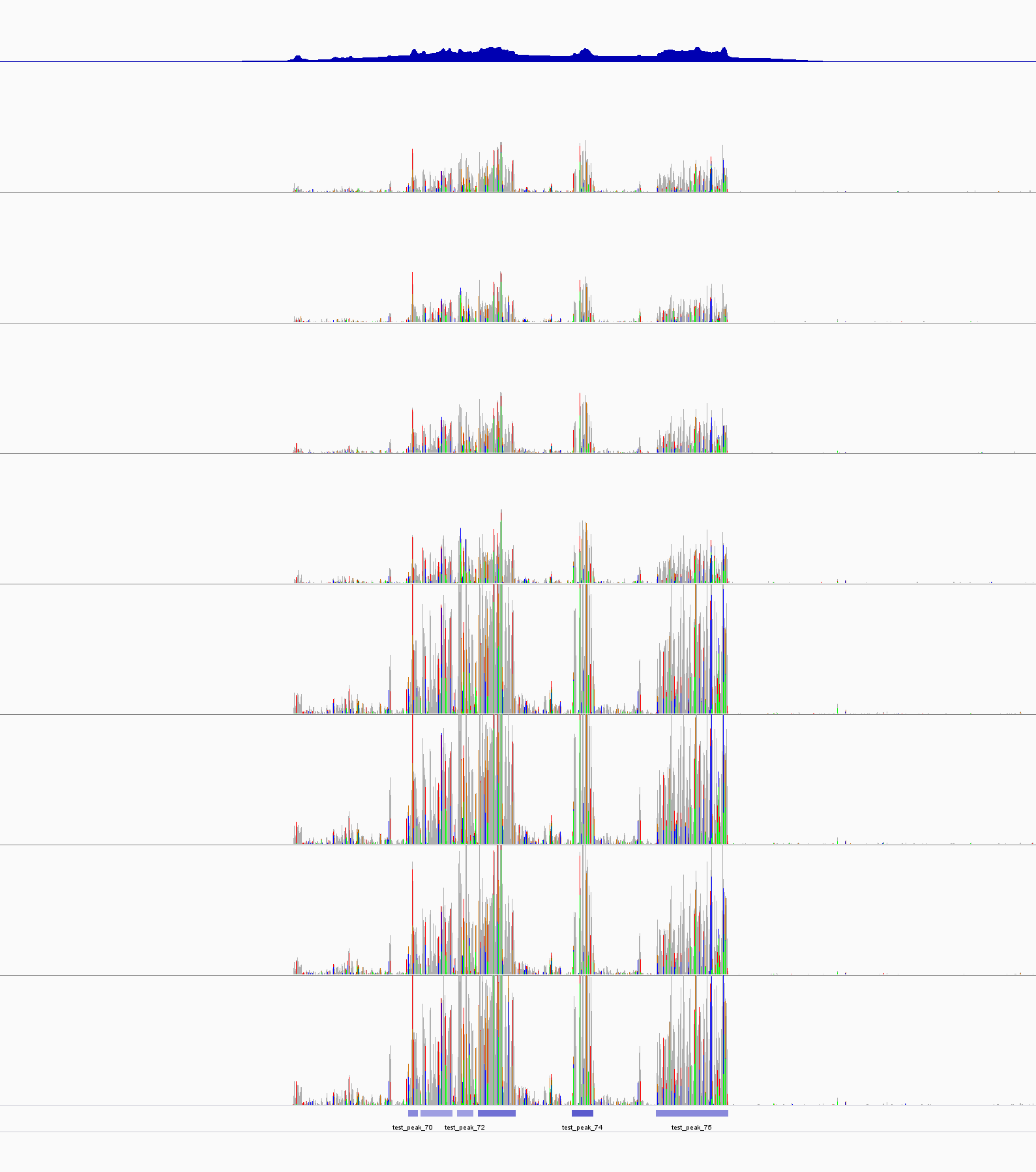 |
|---|
The peak(s) with the best Pvalue. The first track is test_control_lambda.bdg file. the last track is from test_peaks.narrowPeak file. The color of each bar seems corrosponded the P-value. The 5th peak has the lowest p-value and the darkest blue. |
| An example of Unreliable peak. Tow peaks were called based on 3th and 5th but can not be found between 4th and 6th which are biological repeats. |
|---|
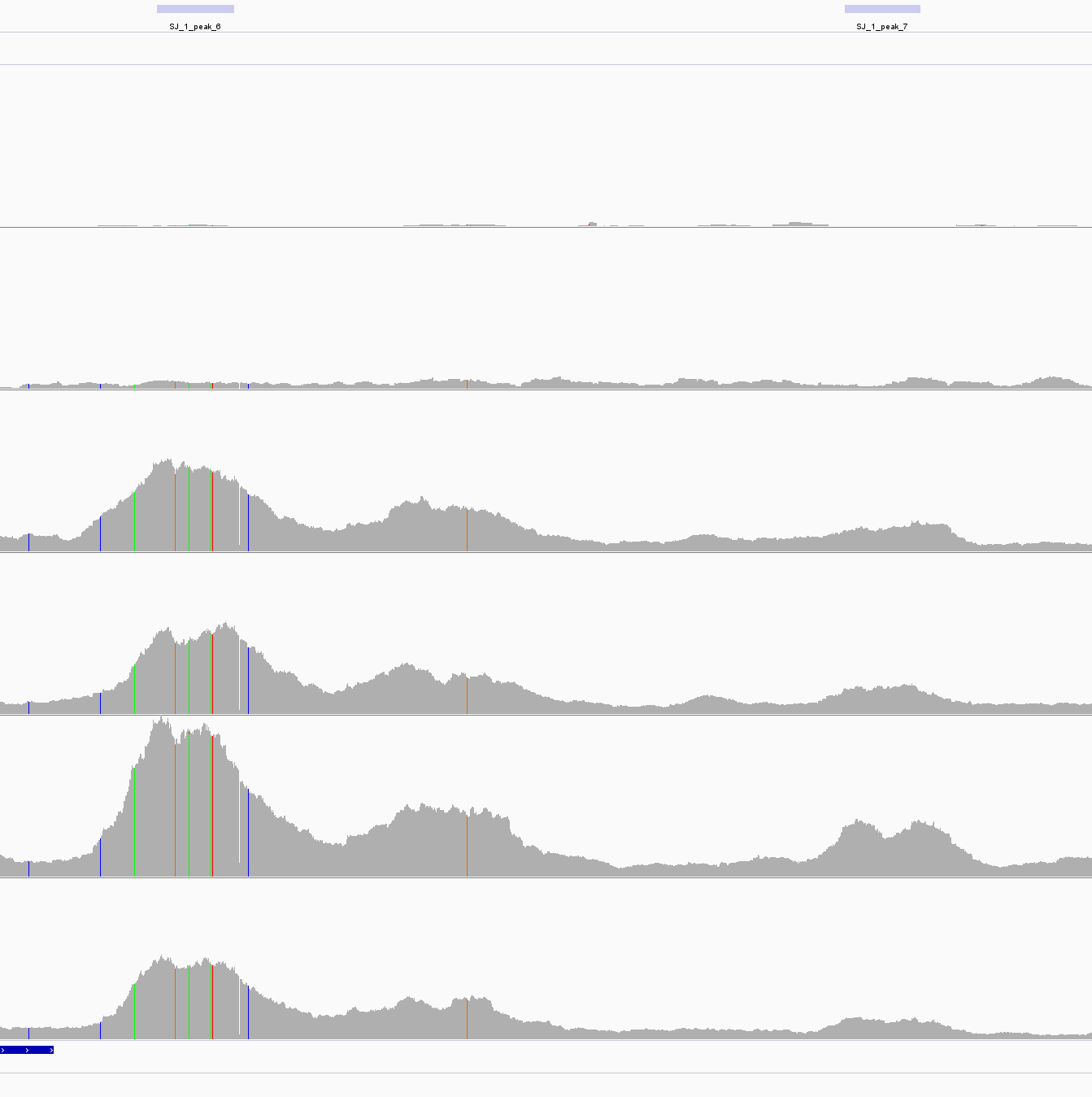 |
ggplot
|
|
After Peak calling
There is a pipeline
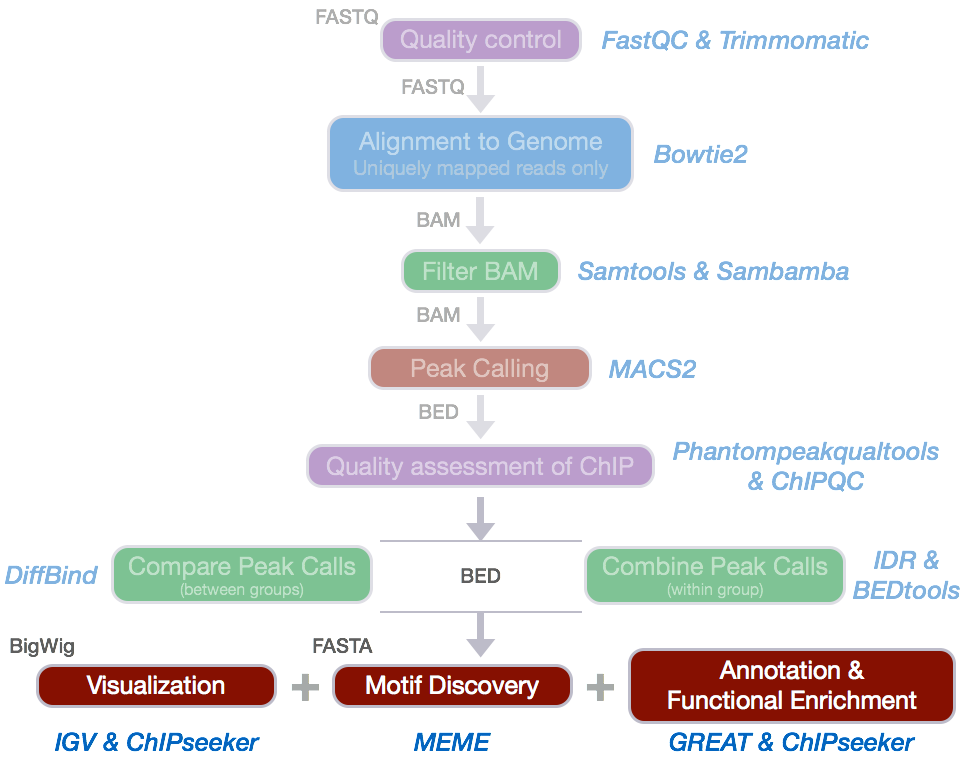
|
hello wold
Chip-Seq, from Know to Unknown = =







