DNA Damage
DNA Damage
DNA is vulnerable to damage resulting from endogenous metabolites, environmental and dietary carcinogens, some anti-inflammatory drugs, and genotoxic cancer therapeutics. Cells respond to DNA damage by activating complex signalling networks that decide cell fate, promoting not only DNA repair and survival but also cell death. The decision between cell survival and death following DNA damage rests on factors that are involved in DNA damage recognition, and DNA repair and damage tolerance, as well as on factors involved in the activation of apoptosis, necrosis, autophagy and senescence. The pathways that dictate cell fate are entwined and have key roles in cancer initiation and progression. Furthermore, they determine the outcome of cancer therapy with genotoxic drugs. Understanding the molecular basis of these pathways is important not only for gaining insight into carcinogenesis, but also in promoting successful cancer therapy. In this Review, we describe key decision-making nodes in the complex interplay between cell survival and death following DNA damage.
© Wynand P. Roos; 2016
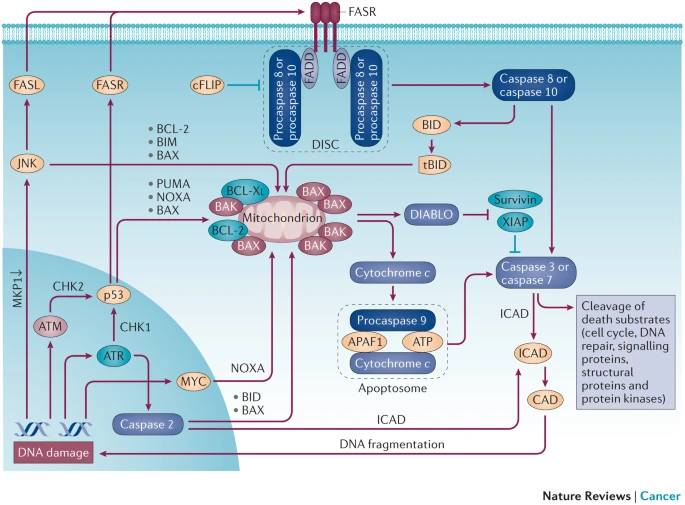
In the event of DNA damage, cells activate DNA repair pathways to facilitate the removal of replication barriers. Conversely, in instances of irreparable DNA damage, cells are prompted to undergo programmed cell death[1].
Reactive oxygen species (ROS), a by-product of regular cellular metabolism, along with various environmental and endogenous genotoxic factors, pose a threat to the stability of DNA and other macromolecules[2].
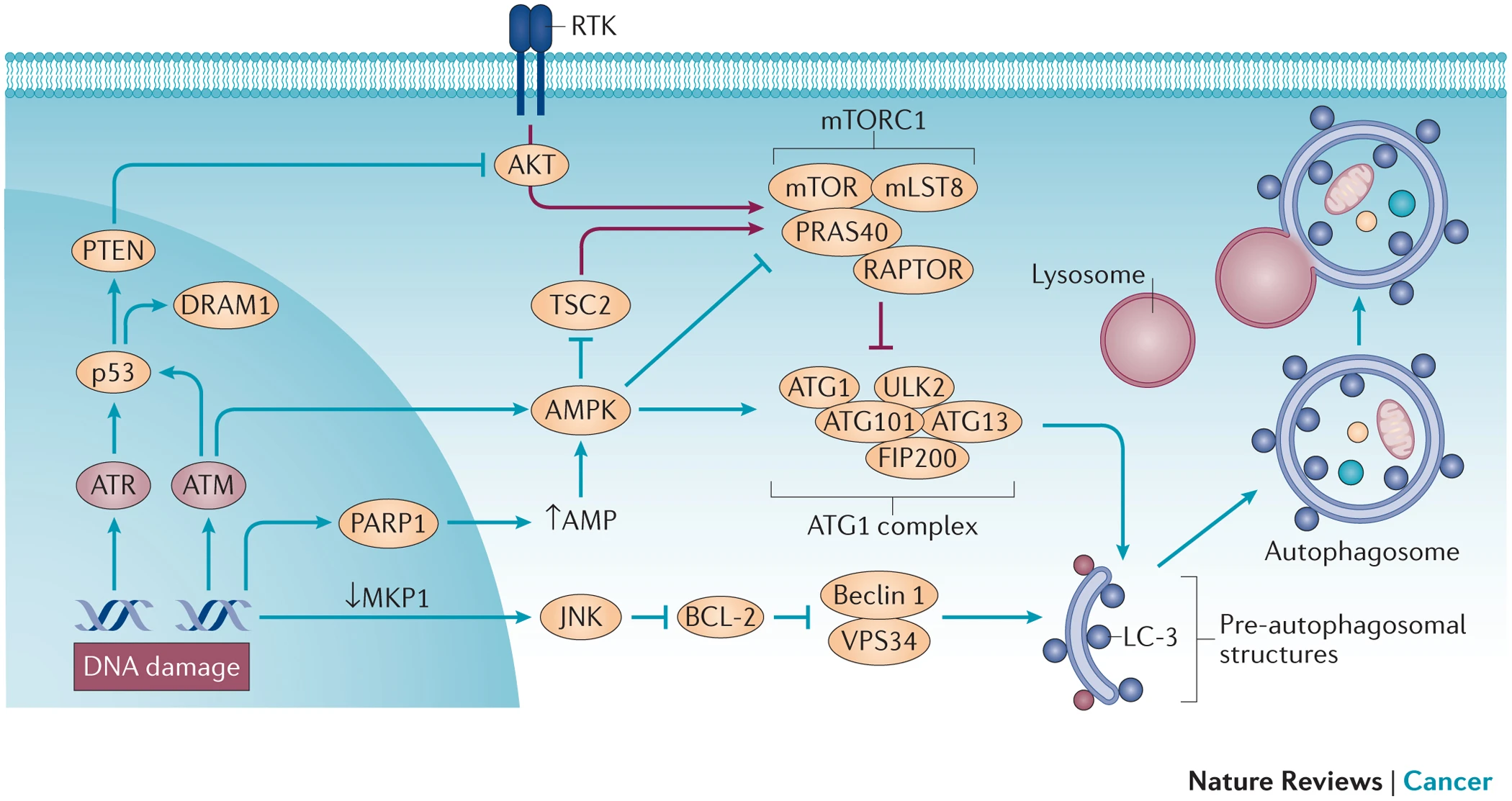
In response to oxidative DNA damage, cells activate cell cycle checkpoints, leading to the arrest of the cell cycle and cessation of DNA replication. This creates a time window during which DNA repair pathways, such as excision repair, can effectively identify and repair DNA damage induced by oxidative stress. Excision repair is a critical system for repairing oxidative stress-induced DNA damage[3].
 |
|---|
| © Wynand P. Roos; 2016 |
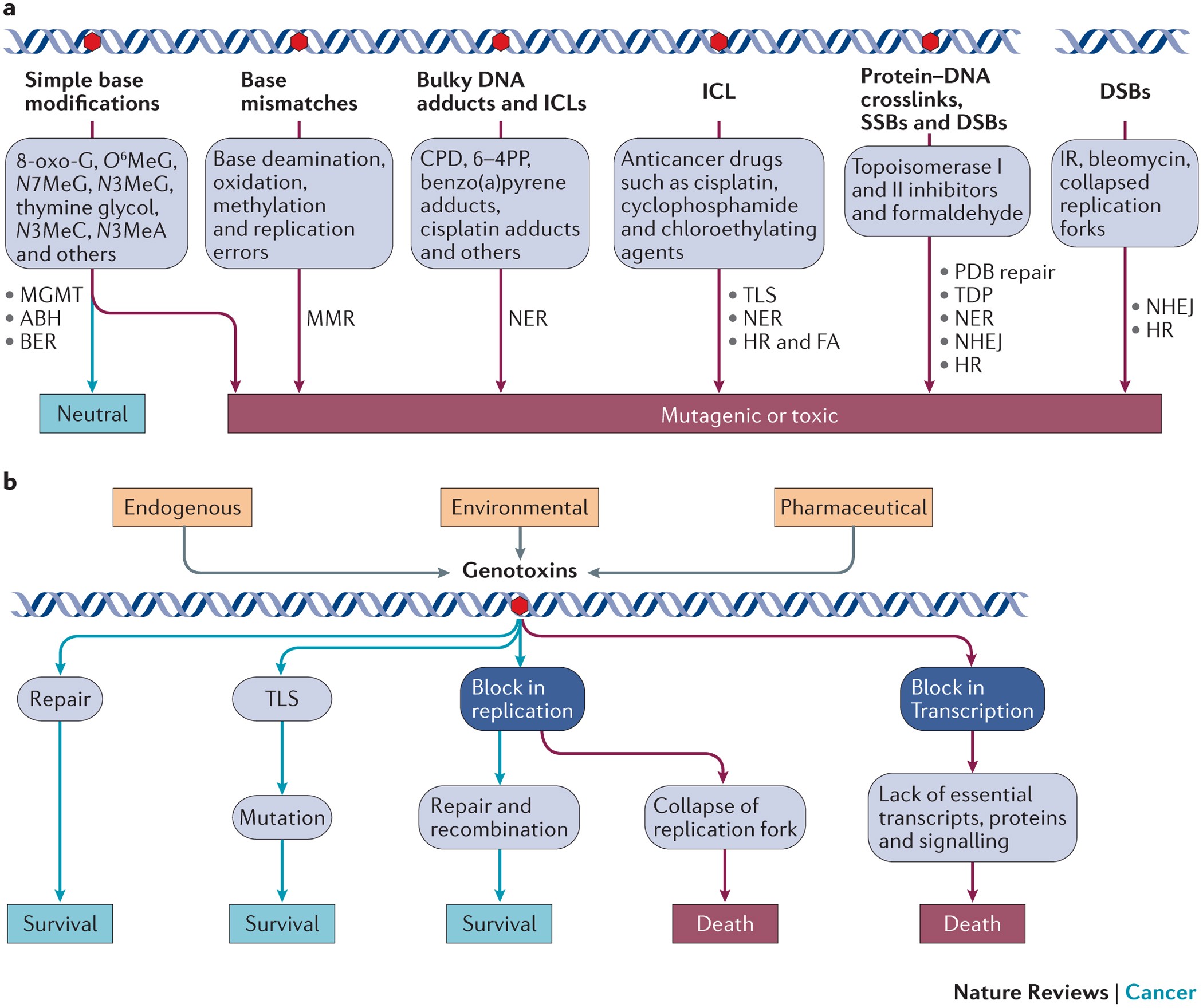
Key damage tolerance mechanisms for cell survival[4][5][6]:
- non-homologous end joining (NHEJ)
- homologous recombination (HR), MMR, BER, nucleotide excision repair (NER)
- protein-linked DNA break (PDB) repair in combination with DDR signallin.
Three immediate-early sensors in the DDR - PI3K-related kinases (PIKKs)[7]:
- ataxia telangiectasia mutated (ATM)
- ataxia telangiectasia and Rad3-related (ATR)
- functionally compromised ATR also show malignancy
- DNA-dependent protein kinase (DNA-PK)
ATM and ATR promote cell death in instances of high DSB levels[13]
 |
|---|
| © Wynand P. Roos; 2016 |
L.H. Pearl, A.C. Schierz, S.E. Ward, B. Al-Lazikani, F.M. Pearl
Therapeutic opportunities within the DNA damage response
Nat. Rev. Cancer, 15 (3) (2015), pp. 166-180 ↩︎M. Majidinia, B. Yousefi DNA damage response regulation by MicroRNAs as a therapeutic target in cancer
DNA Repair, 47 (2016), pp. 1-11 ↩︎Davies, Kelvin JA, ed. Oxidative damage & repair: Chemical, biological and medical aspects. Elsevier, 2013. ↩︎
Roos, W. P. et al. The translesion polymerase Rev3L in the tolerance of alkylating anticancer drugs. Mol. Pharmacol. 76, 927–934 (2009). ↩︎
Ashour, M. E., Atteya, R. & El-Khamisy, S. F. Topoisomerase-mediated chromosomal break repair: an emerging player in many games. Nat. Rev. Cancer 15, 137–151 (2015). ↩︎
Stingele, J., Habermann, B. & Jentsch, S. DNA–protein crosslink repair: proteases as DNA repair enzymes. Trends Biochem. Sci. 40, 67–71 (2015). ↩︎
Rogakou, E. P., Pilch, D. R., Orr, A. H., Ivanova, V. S. & Bonner, W. M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273, 5858–5868 (1998) ↩︎
Stankovic, T. et al. ATM mutations in sporadic lymphoid tumours. Leuk. Lymphoma 43, 1563–1571 (2002). ↩︎
Kim, H. et al. Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clin. Cancer Res. 20, 1865–1872 (2014). ↩︎
Swift, M., Morrell, D., Massey, R. B. & Chase, C. L. Incidence of cancer in 161 families affected by ataxia-telangiectasia. N. Engl. J. Med. 325, 1831–1836 (1991). ↩︎
Thompson, D. et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J. Natl Cancer Inst. 97, 813–822 (2005). ↩︎
Khanna, K. K. Cancer risk and the ATM gene: a continuing debate. J. Natl Cancer Inst. 92, 795–802 (2000). ↩︎
Pusapati, R. V. et al. ATM promotes apoptosis and suppresses tumorigenesis in response to Myc. Proc. Natl Acad. Sci. USA 103, 1446–1451 (2006). ↩︎







