Protein Loop Refinement
DaReUS-Loop 2018 (Web Server Only)
 |
|---|
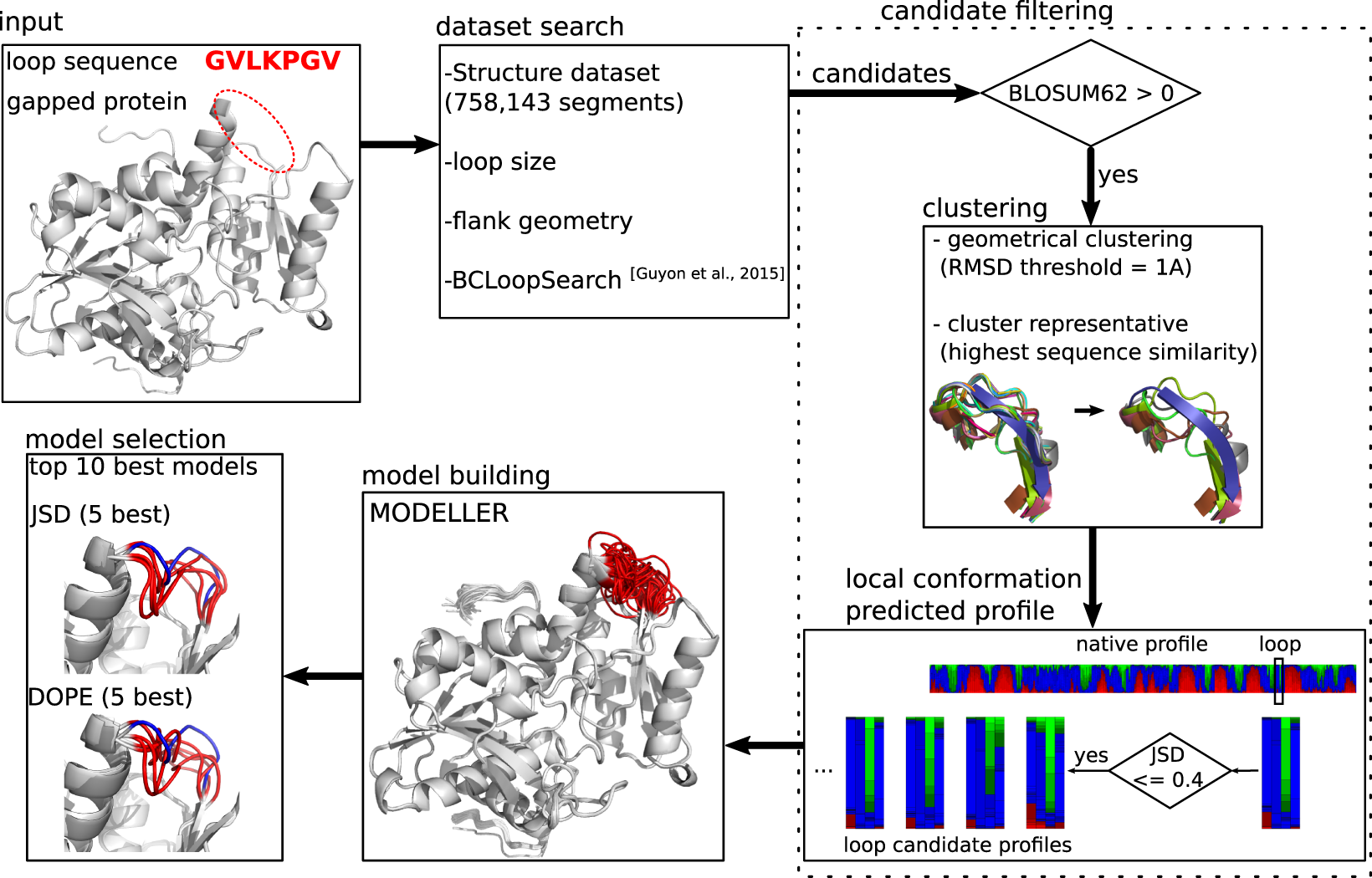
DaReUS-Loop (Data-based approach using Remote or Unrelated Structures for Loop modeling) (Web Server) is a data-based approach that identifies loop candidates mining the complete set of experimental structures available in the Protein Data Bank. Candidate filtering relies on local conformation profile-profile comparison, together with physico-chemical scoring[1]. This work is the extension of using Binet-Cauchy kernel to mine large collections of structures efficiently[2].
The database they used: CASP11, CASP12 and HOMSTRAD.
In this paper, They classified protein loop-modeling tools are generally into three main categories:
- Knowledge-based (Template-based) Methods:
- These approaches utilize structural repositories to extract observed loop conformations that match specific sequences and geometric constraints. By mining databases of known protein structures, they identify loop candidates that fit the target region, offering computational efficiency. However, their effectiveness is limited by the availability of suitable loop conformations in existing structural data.
- Exp: SuperLooper2[3] mines the Loop In Protein (LIP) database;
- MoMA-LoopSampler[4]: a knowledge-based method that uses a database of loop fragments to model missing segments in proteins. It has web server but queue is too long to wait.
- Ab Initio (De Novo) Methods:
- These techniques involve sampling a wide range of possible loop conformations without relying on existing structural templates. They dependent on energy optimization techniques and are consequently highly time consuming. They often employ exhaustive searches of the loop’s torsional angles to explore conformational space. While capable of modeling loops without suitable templates, ab initio methods are computationally intensive and typically more successful with shorter loops due to the vast number of possible conformations.
- Exp:Rosetta Next-Generation KIC (NGK)[5], GalaxyLoop-PS2[6]
- Hybrid Methods:
- Combining elements of both knowledge-based and ab initio approaches, hybrid methods use small structural fragments from databases within an ab initio sampling framework. This integration aims to balance computational efficiency with modeling accuracy, leveraging known structural motifs to guide the exploration of conformational space.
- Exp: Sphinx[7]
How to Use
In the Web Server, you can upload your protein structure file and the sequences.
For the sequences, you need to mask your loop with gap (‘-’).
In my case, I masked a 14 amino acid loop in the sequence and this is the log:
[00:00:00] Please bookmark this page to have access to your results! [00:00:00] 1/14: Verifying input files [00:00:00] Warning! Only the first chain from the input PDB is considered! [00:00:11] 2/14: BCLoopSearch [00:01:42] 3/14: clustering [00:01:59] 4/14: Conformational profiles [00:04:16] Found 353 hits for Loop1_AMTMVVASFFQYYA [00:04:16] 5/14: Measuring Jensen Shannon distances 10%..20%..30%..40%..50%..60%..70%..80%..90%..100% [00:08:06] 6/14: selecting top candidates per loop [00:08:21] 7/14: preparing top candidates for minimization [00:08:43] 8/14: positioning linker side chains [00:09:36] 9/14: minimization [00:11:01] 10/14: Scoring the candidates [00:11:01] ... 3 jobs already in the queue, please wait ... [00:23:16] 11/14: Measuring energy values (KORP potential) [00:23:45] 12/14: Preparing the final energy report [00:23:55] 13/14: Generating combinatorial models [00:24:04] 14/14: detecting possible clashes between candidates [00:24:34] ... 3 jobs already in the queue, please wait ... [00:26:46] ... 2 jobs already in the queue, please wait ... [00:27:52] Finished

In the end, you can download the top 10 models.
I compared it with the original structure and the loop is refined and find none of them are making sense. Another biggest problem is that they only took the chain with masked loops and disgard the rest of other chains. In this case, lots of results would have a serious atom crush problem. So, it takes about half an hour to get the result and the result is not good even for a very small protein.
By checking the performance from the paper, it also doesn’t show a dramatic improvements.
Foldseek 2024
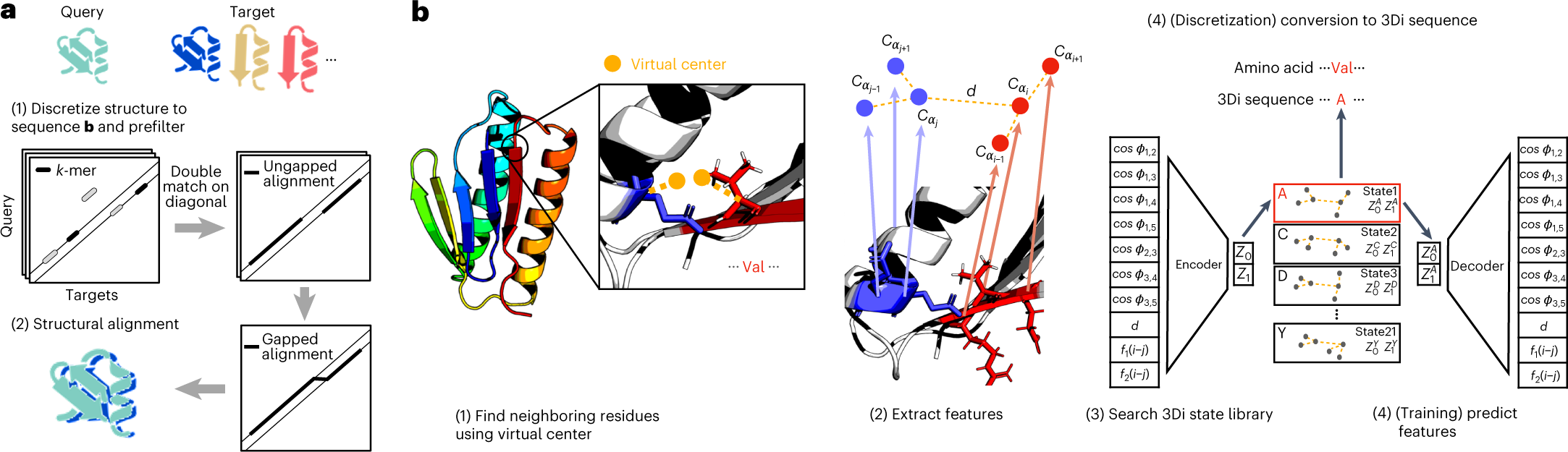
FOldSeek[8] also has a web server. It also deposited there codes on Github at steineggerlab/foldseek. After that, the also development foldseek-multimer[9] for protein complex alignment.
Abstract
As structure prediction methods are generating millions of publicly available protein structures, searching these databases is becoming a bottleneck. Foldseek aligns the structure of a query protein against a database by describing tertiary amino acid interactions within proteins as sequences over a structural alphabet. Foldseek decreases computation times by four to five orders of magnitude with 86%, 88% and 133% of the sensitivities of Dali, TM-align and CE, respectively.It is a fast loop searching tool which is very powerful if you want to use it to search the loop in a large database. It is a very good tool if you want to development a new knowledge-based loop refinement tool. You can’t use it to refine the loop directly.
KarmaLoop 2024
 |
|---|
KarmaLoop[10] is an open source deep learning-based framework designed for rapid and precise full-atom protein loop modeling. It addresses the challenges of predicting loop conformations, which are critical for accurate protein structure determination.
Key Features of KarmaLoop:
-
Full-Atom Modeling: Unlike many methods that focus primarily on backbone atoms, KarmaLoop predicts the positions of both backbone and side-chain heavy atoms, providing comprehensive loop conformations.
-
Deep Learning Architecture: The framework employs advanced neural network architectures, including Graph Transformers and Geometric Vector Perceptrons, to effectively capture intricate structural patterns within proteins.
-
High Accuracy and Efficiency: KarmaLoop has demonstrated superior performance in both accuracy and computational speed compared to traditional and other deep learning-based methods. For instance, it achieved average root-mean-square deviations (RMSDs) of 1.77 Å and 1.95 Å on the CASP13+14 and CASP15 benchmark datasets, respectively, with a significant speed advantage over other methods. citeturn0search0
-
Versatility: The tool has shown effectiveness in modeling general protein loops as well as specific regions like antibody complementarity-determining region (CDR) H3 loops, which are known for their structural complexity.
How to Use
It is very easy to installing.
Before you refine you loop, you need to truncate you structure with the script they provide and convert it into graphic. Then, you can use their pre-trained model to refine your structure.
The problem is the script to make the graphic has different features on the model the give you. So, you can use their model, at all. It seems they updated the codes for model alone, but forget to update the codes for graphic making. Someone asked this question on the github but didn’t get any response.
PLM-GAN
PLM-GAN looks like a good model. But they deposited their trained model on github as a large file and we can’t download it any more. So, we can’t test the performace of it.
It applies Generative Adversarial Networks (GANs) to model loop structures, leveraging the generator-discriminator framework to produce realistic loop conformations. It utilizes the pix2pix GAN model to generate and inpaint protein distance matrices, facilitating the folding of protein structures
Karami Y, Guyon F, De Vries S, et al. DaReUS-Loop: accurate loop modeling using fragments from remote or unrelated proteins[J]. Scientific reports, 2018, 8(1): 13673. ↩︎
Guyon F, Tufféry P. Fast protein fragment similarity scoring using a binet–cauchy kernel[J]. Bioinformatics, 2014, 30(6): 784-791. ↩︎
Ismer J, Rose A S, Tiemann J K S, et al. SL2: an interactive webtool for modeling of missing segments in proteins[J]. Nucleic acids research, 2016, 44(W1): W390-W394. ↩︎
MoMA-LoopSampler: a web server to exhaustively sample protein loop conformations ↩︎
Stein A, Kortemme T. Improvements to robotics-inspired conformational sampling in rosetta[J]. PloS one, 2013, 8(5): e63090. ↩︎
Park H, Lee G R, Heo L, et al. Protein loop modeling using a new hybrid energy function and its application to modeling in inaccurate structural environments[J]. PloS one, 2014, 9(11): e113811. ↩︎
Marks C, Nowak J, Klostermann S, et al. Sphinx: merging knowledge-based and ab initio approaches to improve protein loop prediction[J]. Bioinformatics, 2017, 33(9): 1346-1353. ↩︎
Van Kempen M, Kim S S, Tumescheit C, et al. Fast and accurate protein structure search with Foldseek[J]. Nature biotechnology, 2024, 42(2): 243-246. ↩︎
Kim W, Mirdita M, Levy Karin E, et al. Rapid and sensitive protein complex alignment with foldseek-multimer[J]. Nature Methods, 2025: 1-4. ↩︎
Wang T, Zhang X, Zhang O, et al. Highly accurate and efficient deep learning paradigm for full-atom protein loop modeling with KarmaLoop[J]. Research, 2024, 7: 0408. ↩︎
Protein Loop Refinement
https://karobben.github.io/2025/03/03/Bioinfor/protein-loop/







