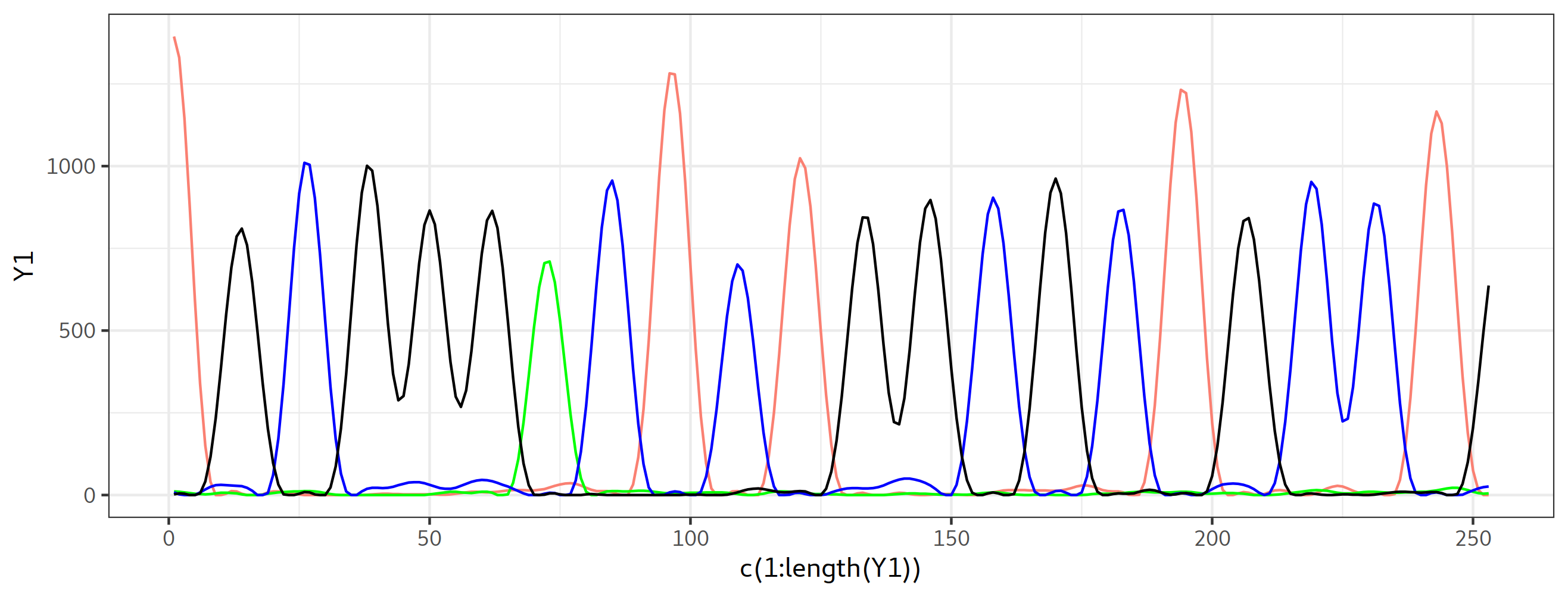
library(ggplot2)
Y1 = head(A@data$DATA.9,3000)
Y2 = head(A@data$DATA.10,3000)
Y3 = head(A@data$DATA.11,3000)
Y4 = head(A@data$DATA.12,3000)
ggplot() + geom_path(aes(x=c(1:length(Y1)), y= Y1),color='salmon')+
geom_path(aes(x=c(1:length(Y2)), y= Y2),color='green')+
geom_path(aes(x=c(1:length(Y3)), y= Y3),color='blue')+
geom_path(aes(x=c(1:length(Y4)), y= Y4),color='black')+
theme_bw()
ABI_plot <- function(A, Head=0, Tail=1000, alpha = 0.7, Type="Base"){
if(Type!="QS"){
if(Type=="Base"){
Y1 = tail(head(A@data$DATA.9, Tail),Tail-Head)
Y2 = tail(head(A@data$DATA.10, Tail),Tail-Head)
Y3 = tail(head(A@data$DATA.11, Tail),Tail-Head)
Y4 = tail(head(A@data$DATA.12, Tail),Tail-Head)
}
if(Type=="Raw"){
Y1 = tail(head(A@data$DATA.1, Tail),Tail-Head)
Y2 = tail(head(A@data$DATA.2, Tail),Tail-Head)
Y3 = tail(head(A@data$DATA.3, Tail),Tail-Head)
Y4 = tail(head(A@data$DATA.4, Tail),Tail-Head)
}
P <- ggplot() +
geom_path(aes(x=c((Head+1):Tail), y= Y1, color = "G"), alpha = alpha)+
geom_path(aes(x=c((Head+1):Tail), y= Y2, color = "A"), alpha = alpha)+
geom_path(aes(x=c((Head+1):Tail), y= Y3, color = "T"), alpha = alpha)+
geom_path(aes(x=c((Head+1):Tail), y= Y4, color = "C"), alpha = alpha)+
theme_bw() +
scale_color_manual(name = "Group",
values = c( "A" = "green", "T" = "salmon", "G" = "black", "C" = "blue"),
labels = c("A", "T", "G","C"))
}
if(Type=="QS"){
Y1 = tail(head(A@data$PCON.1, Tail),Tail-Head)
Y2 = tail(head(strsplit(A@data$PBAS.1, "", perl = TRUE)[[1]], Tail),Tail-Head)
P <- ggplot() +
geom_bar(aes(x=c((Head+1):Tail), y= Y1, fill= Y1), stat = "identity", alpha =1)+
scale_fill_gradient(low = "white", high = "Tomato3", limits = c(0,62)) +
theme_bw()+
geom_text(aes(x=c((Head+1):Tail), y= -6, label = Y2))
}
print(P)
}
BS = length(A@data$DATA.9) / length(A@data$PCON.2)
ABI_plot(A, BS*100, BS*120)
|