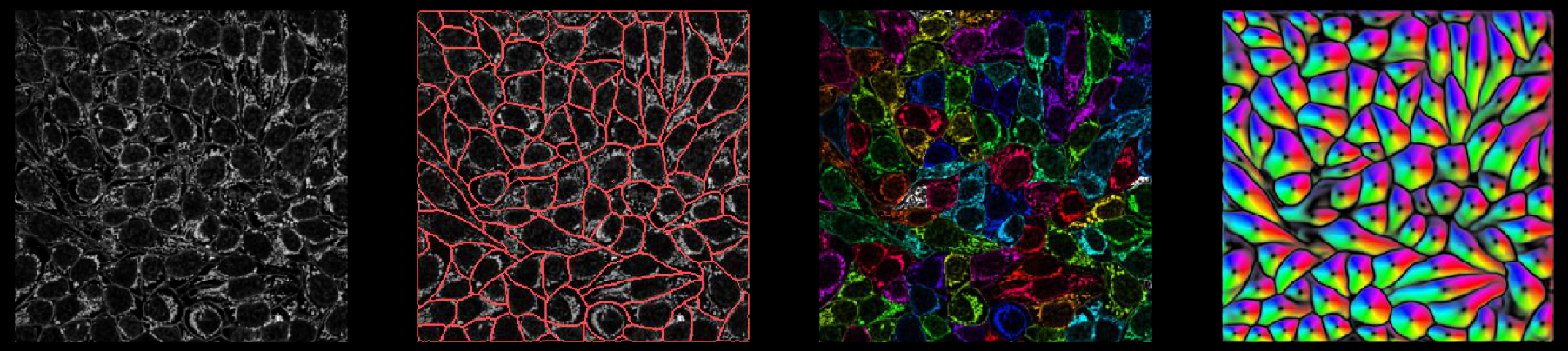
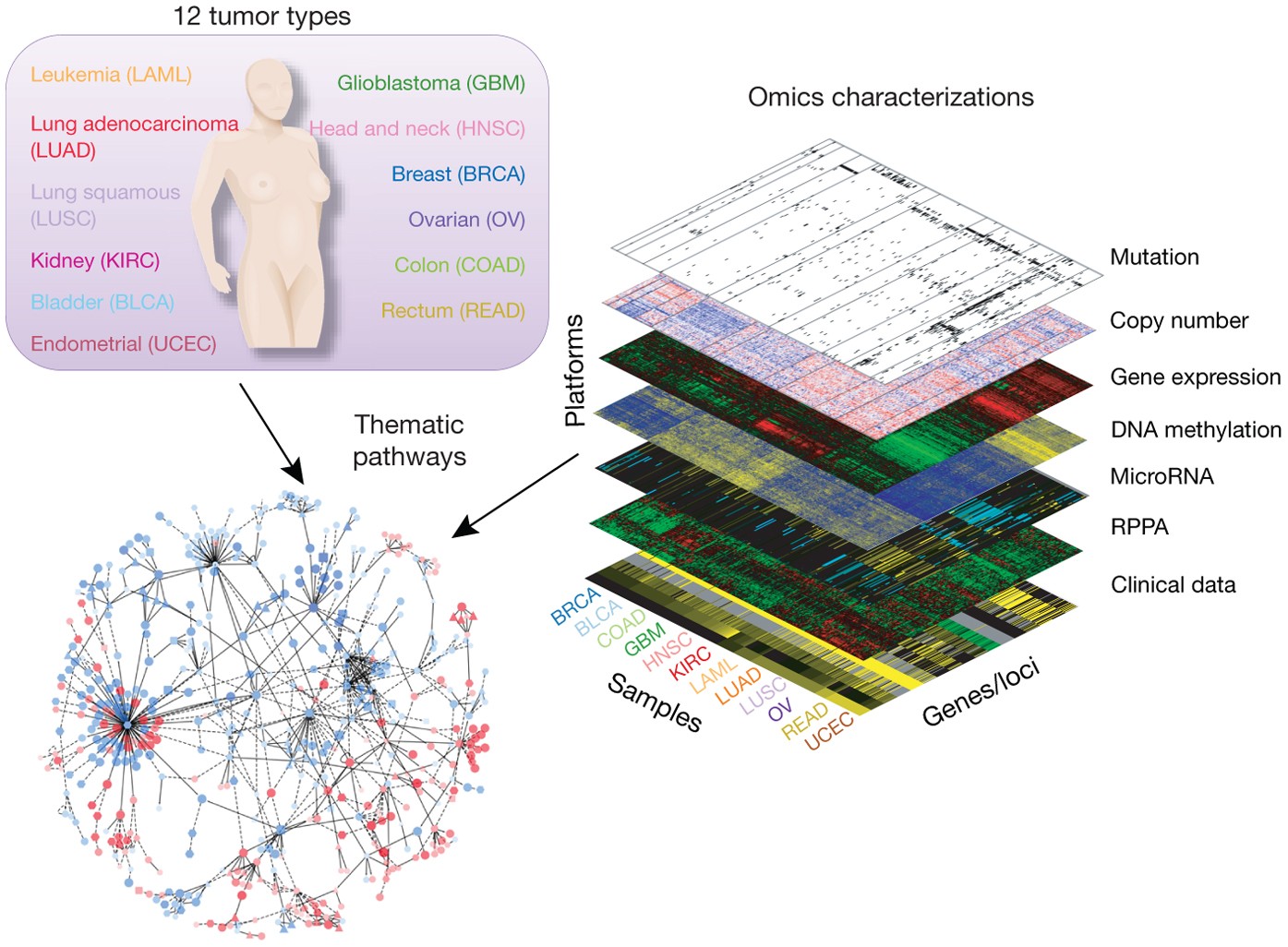
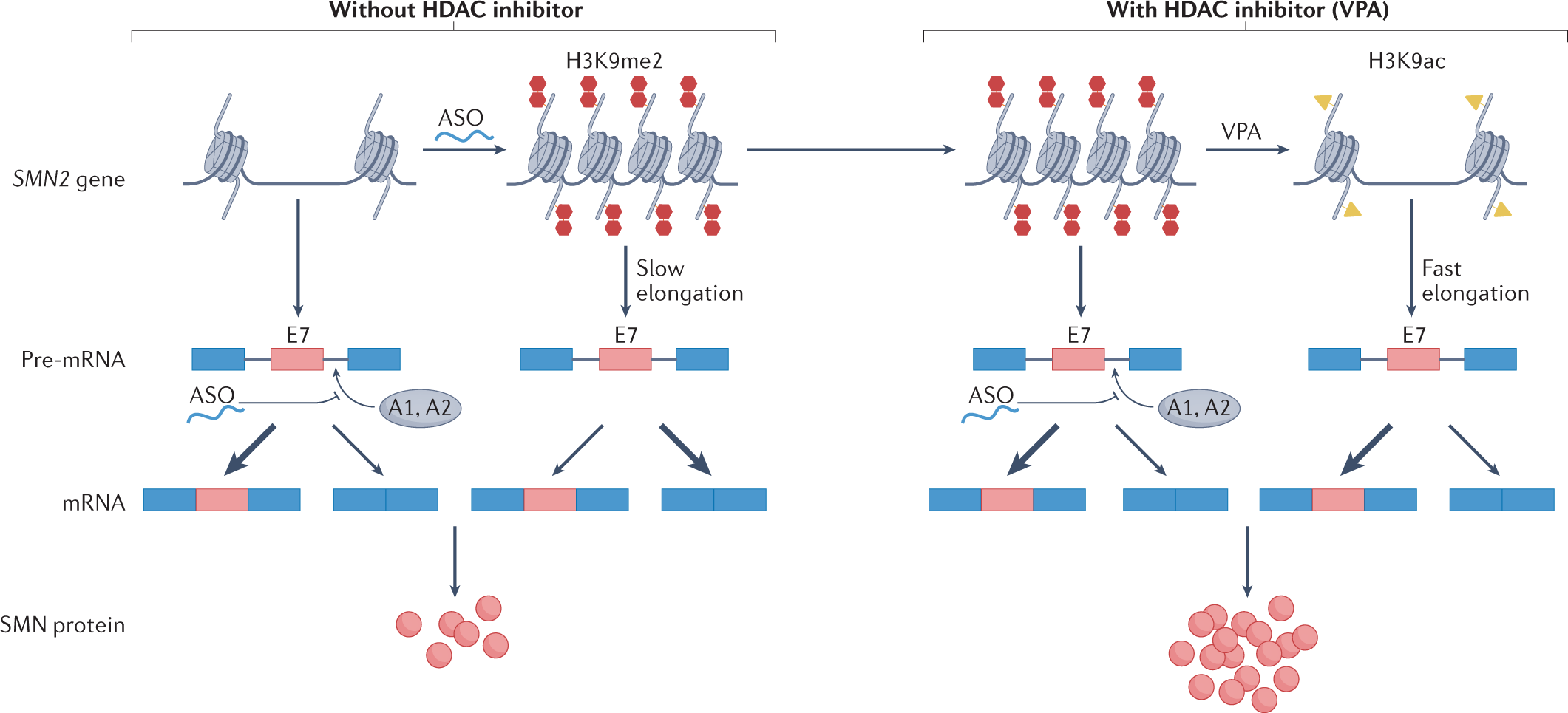
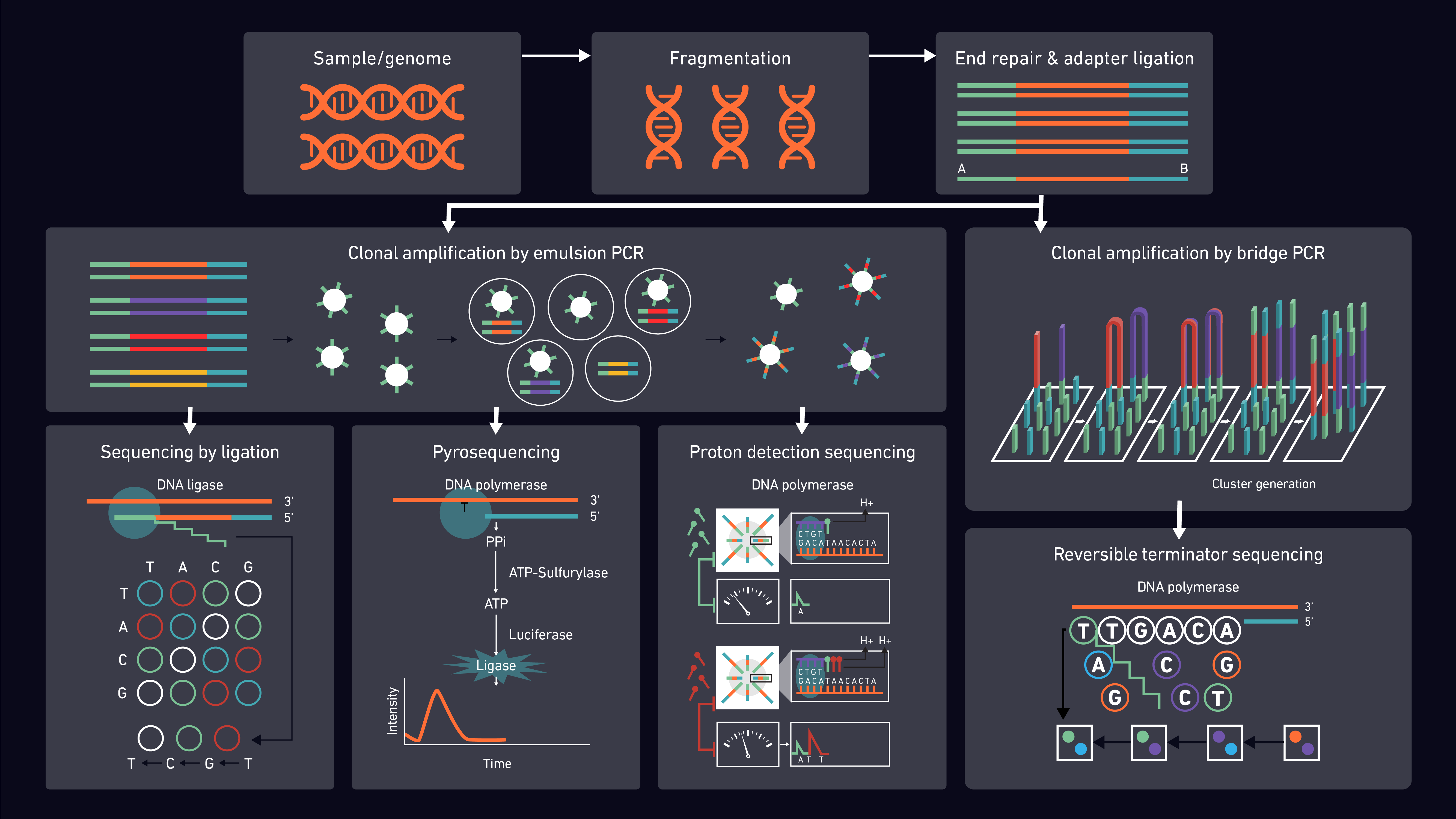
Behavior Analysis
Behavioral research is a vast field that offers insights into the complexities of behavior and provides a foundation for making predictions and interventions in various sectors of life and society.
Read more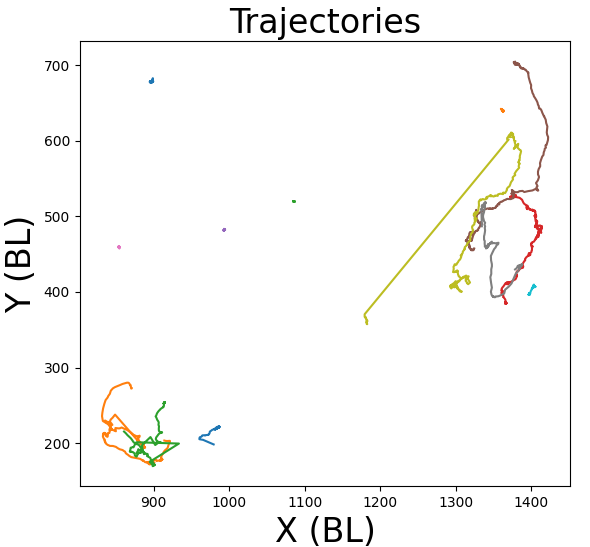 © Karobben
© Karobben © Karobben
© Karobben © Karobben
© Karobben © Karobben
© Karobben © Karobben
© Karobben