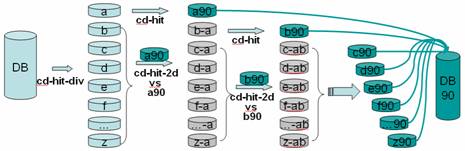
CD-HIT was originally a protein clustering program. The main advantage of this program is its ultra-fast speed. It can be hundreds of times faster than other clustering programs, for example, BLASTCLUST. Therefore it can handle very large databases, like NR. The 1st version of this program, CD-HI, was published and released in 2001. The 2nd version, called CD-HIT, was published in 2002 with significant improvements. Since 2004, CD-HIT has been hosted at bioinformatics.org as an open source project. Current CD-HIT package can perform various jobs like clustering a protein database, clustering a DNA/RNA database, comparing two databases (protein or DNA/RNA), generating protein families, and many others.
Read more